
Abstract
Rapid gonad development is an important concern in the Macrobrachium nipponense aquaculture industry. The process of sexual differentiation and development is influenced by environmental factors, including water temperature and illumination time. In this study, we aimed to identify the key metabolites and genes from the androgenic gland, in response to different water temperatures and illumination times based on the integrated metabolomic and transcriptomic analysis of the androgenic gland during the reproductive vs. non-reproductive seasons. Histological observations revealed that the division of the androgenic gland in the reproductive season is more active than that in the non-reproductive season. The metabolic profiling analysis revealed that glycerophospholipid metabolism and sphingolipid metabolism represented the most enriched metabolic pathways of differentially expressed metabolites (DEMs). Transcriptome profiling analysis revealed that phagosome, spliceosome, RNA degradation, ribosome biogenesis in eukaryotes, and oxidative phosphorylation represented the main enriched metabolic pathways of differentially expressed genes (DEGs). A total of 14 DEGs from these metabolic pathways were considered as strong candidate genes involved in the male sexual development in M. nipponense, based on the expression difference between the reproductive vs. non-reproductive seasons. qPCR verifications of these 14 DEGs indicate the accuracy of the RNA-Seq. In addition, eight genes out of 14 DEGs showed high expression levels in the testis or/and androgenic gland. Predicting these eight genes may play an essential role in male sexual development of M. nipponense. The results of this study contribute to our knowledge of sexual development in male M. nipponense, as well as other crustacean species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The oriental river prawn Macrobrachium nipponense has a great market value in China and other Asian countries (Fu et al. 2012). The annual production of M. nipponense reached 205,010 tons in 2020, making it an economically important species (Zhang et al. 2020). Male M. nipponense grow faster and reach larger sizes (approximately 2–2.5 times larger) at harvest time, compared to the female counterparts. Thus, production of the all-male population provides economic benefits for the M. nipponense culture industry. In addition, the rapid gonad development of newly hatched prawn larvae during the reproductive season is the main problem in the M. nipponense culture industry. Both the testis and ovaries of the hatchlings developed to sexual maturity within 40 days after hatching. This causes inbreeding between young prawns. Inbreeding leads to the mating and propagation of multiple generations in the same ponds, resulting in the prawns with smaller market size, restricting the sustainable development of the M. nipponense industry (Jin et al. 2021a, b, c). Therefore, the identification of important sex-determining and reproduction-related metabolites and genes are urgently needed, as it is integral in the establishment of an artificial technique to produce all progenies on a commercial scale and to regulate the process of gonad development in M. nipponense.
The androgenic gland is a unique endocrine organ in male crustaceans. The androgenic gland and the hormones it secretes have been reported to play an essential role in controlling sex differentiation and maintaining male secondary sexual characteristics in crustaceans, especially in testis development (Sagi et al. 1986, 1990). Insulin-like androgenic gland hormone (IAG) is the main expressed gene in the androgenic gland, which was reported to be the most important sex-related gene in crustacean species (Rosen et al. 2010; Ventura et al. 2009, 2011). Knocking down the expression of IAG resulted in the sex-reversal in Macrobrachium resenbergii (Ventura et al. 2012). IAG also plays essential roles in the mechanism of male sexual differentiation and development in M. nipponense (Ma et al. 2016), Fenneropenaeus chinensis (Li et al. 2012), Scylla paramamosain (Huang et al. 2014), Lysmata vittate (Liu et al. 2021), and Fenneropenaeus merguiensis (Zhou et al. 2021). Moreover, the mechanism of male sexual differentiation and development in M. nipponense, based on the androgenic gland, has been successfully determined (Jin et al. 2013; Jin et al. 2018; Jin et al. 2019a, b; Ma et al. 2016). However, some important male reproduction-related metabolites and genes of M. nipponense are still unclear, which hinders the establishment of an artificial technique to regulate the process of testis development.
Previous studies reported that temperature, illumination, and the presence of chemical pollutants dramatically affect the process of sexual differentiation and development in aquatic animals, leading to sex reversal (Wedekind 2017). Environmental factors can influence the expression profiles of DMRT1 and vitellogenin, which affect the sexual differentiation and development of lower vertebrates (Leslie and Valentine 2015; Om et al. 2015). The reproductive season of M. nipponense is from April to October in a year with the water temperature ≥ 28 °C and illumination time of ≥ 16 h, while the optimal period is from June to July. The non-reproductive season of M. nipponense is from December to February of the next year with the water temperature ≤ 15 °C and illumination time of ≤ 10 h (Fu et al. 2012). Thus, the identification of metabolites and genes from the androgenic gland, in response to water temperature and illumination time, play essential roles to fully understand the mechanism of sexual differentiation and development in M. nipponense.
In this study, we aimed to identify the morphological differences of the androgenic gland during the reproductive season in July (water temperature ≥ 28 °C and illumination time ≥ 16 h) vs. that during the non-reproductive season in January (water temperature ≤ 15 °C and illumination time ≤ 10 h) by hematoxylin and eosin (HE) staining. The DEMs and DEGs were selected from the androgenic gland in M. nipponense, in response to different water temperatures and illumination times through the conduct of metabolomic and transcriptomic profiling analysis of the androgenic gland samples taken during the reproductive vs. non-reproductive seasons, which may be involved in the mechanism of sex differentiation and development. This study provided valuable evidences to better understand the molecular mechanisms, underlying sexual differentiation and development in male M. nipponense.
Materials and methods
Sample collection
A total of 200 healthy male prawns with body weights ranging from 3.23 to 5.16 g and 3.45 to 4.98 g were collected from a wild population in Tai Lake, Wuxi, China (120°13′44″E, 31°28′ 22″N) in January (non-reproductive season) and July (reproductive season), respectively. The specimens from the two batches were fed the same commercial diet. An additional 20 healthy female prawns with body weights of 2.76–3.12 g were collected from a wild population in Tai Lake in July. These specimens were maintained for 72 h in a pond with a dissolved oxygen level ≥ 6 mg/L, prior to tissue collection. The water temperature was ≥ 28 °C and illumination time was ≥ 16 h in July, while the water temperature was ≤ 15 °C and illumination time was ≤ 10 h in January. The prepared samples in this study are listed in Table 1. Androgenic glands were each collected from five individual male prawns in January and July, respectively, and maintained in 4% paraformaldehyde until histological analyses. A total of 20 androgenic glands were collected and pooled to form a biological replicate in January and July, respectively, and a total of eleven biological replicates were collected, of which eight replicates were prepared for metabolomic profiling analysis, and three replicates were prepared for the transcriptome profiling analysis. Samples of eight tissues were collected from male prawns (N = 5) in July for qPCR analysis, including the eyestalk, brain, heart, hepatopancreas, gill, muscle, testis, and androgenic gland, while ovaries were collected from female prawns (N = 5) also in July. The tissue samples were immediately frozen in liquid nitrogen and stored at – 80 °C until RNA extraction, in order to prevent RNA degradation.
Histological observations
The morphological differences of the androgenic gland in M. nipponense between the reproductive vs. non-reproductive seasons were determined by HE staining. The detailed procedure for HE staining has been provided in previous studies (Ma et al. 2006; ShangGuan et al. 1991). Briefly, the tissues were first dehydrated by using 50%, 70%, 80%, 95%, and 100% ethanol. The tissues were then made transparent and embedded using alcohol/xylene (1:1), xylene, xylene/wax (1:1), and wax. The embedded tissues were sectioned to a thickness of 5 µm using a slicer (Leica, Wetzlar, Germany) and placed on a slide. The slides were then stained with HE for 3–8 min. The histological slides were observed under an Olympus SZX16 microscope (Olympus Corporation, Tokyo, Japan).
Metabolome profiling analysis
Metabolomic profiling was used to select DEMs from the androgenic gland between the reproductive vs. non-reproductive seasons, which may be involved in the mechanism of male sexual differentiation and development in M. nipponense. The DEMs were determined using liquid chromatography-mass spectrometry analysis (Ortiz-Villanueva et al. 2017). The procedures for metabolomic analysis have been described by Jin et al. (2020). The criterion of the robustness and predictive ability of the model was set as a seven-fold cross-validation, and further validation was performed using permutation tests.
Transcriptome profiling analysis
The DEGs of the androgenic gland between reproductive vs. non-reproductive seasons were identified through transcriptome profiling analysis. The transcriptome profiling analysis was performed on an Illumina High-seq 2500 sequencing platform. Previously published studies have described detailed procedures for RNA-Seq and analysis (Jin et al. 2013, 2021b). Trinity program (version: trinityrnaseq_r20131110) was used to assemble the clean data into non-redundant transcripts (Grabherr et al. 2011). Gene annotation was then performed in the Nr (non-redundant) database, Gene Ontology (GO) (Ashburner et al. 2000), Cluster of Orthologous Groups (COG) (Tatusov et al. 2003), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (Kanehisa et al. 2008), using an E-value of 10−5 (Jin et al. 2013). EB-seq algorithm was used to filter the differentially expressed genes, under the criteria of FDR (False discovery rate) < 0.05 (Benjamini et al. 2001).
qPCR analysis
qPCR was used to verify the accuracy of RNA-Seq and to determine the mRNA expressions of important DEGs in different mature tissues. The detailed procedures have been well described in previously published studies (Jin et al. 2019a, 2021a; Zhang et al. 2013). Briefly, total RNA was extracted from each tissue, using the UNlQ-10 Column Trizol Total RNA Isolation Kit (Sangon, Shanghai, China) following the manufacturer’s protocol. A total of 1 μg total RNA from each tissue was used to synthesize the cDNA template by using the PrimeScript™ RT reagent kit (Takara Bio Inc., Japan). The expression level of each tissue was determined using the UltraSYBR Mixture (CWBIO, Beijing, China). The qPCR analysis was performed on the Bio-Rad iCycler iQ5 Real-Time PCR System (Bio-Rad), and the SYBR Green RT-qPCR assay was used. The primers for qPCR analysis are listed in Table 2. The eukaryotic translation initiation factor 5A (EIF) was used as a reference gene in this study (Hu et al. 2018). The relative expression levels were measured using the 2−ΔΔCT method (Livak and Schmittgen 2001).
Statistical analysis
Quantitative data were expressed as the mean ± SD. Statistical differences were estimated by one-way ANOVA, followed by LSD and Duncan’s multiple range test. All statistical analysis was carried out using SPSS Statistics 23.0. A probability level of 0.05 was used to indicate significance (p < 0.05).
Results
Histological observations of the androgenic gland
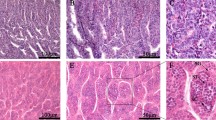
Histological observations revealed that the androgenic gland taken during the non-reproductive vs. reproductive seasons showed significant morphological differences (Fig. 1). The division of the androgenic gland during the reproductive season was more active than that during the non-reproductive season. The androgenic gland cells in the non-reproductive season were smaller than that during the reproductive season. The cell boundary was clear, and the cytoplasm was basophilic. The nucleolus of the nucleus was not visible in the non-reproductive season. However, the androgenic gland cells were loose in the reproductive season. The intercellular connections were closer in the reproductive season, and multiple nucleoli were observed in the nucleus.

Morphological difference of androgenic gland between reproductive season vs. non-reproductive season. AGC androgen gland cell, TA ampulla, AG androgenic gland
Metabolic profiling analysis
The overall quality of the metabolic profiling analysis of the androgenic gland between the non-reproductive vs. reproductive seasons was measured by principal component analysis (Fig. 2) and latent structures discriminant analysis (Fig. 3). The R2X (cum), R2Y (cum), and Q2 (cum) were 0.942, 1, and 0.947, respectively. The R2 and Q2 intercept values were 0.943 and − 0.906 after 200 permutations of the treatments. A total of 260 DEMs were identified from the androgenic gland between the non-reproductive vs. reproductive seasons, of which 148 DEMs were upregulated and 112 DEMs were downregulated in the non-reproductive season, using the criteria of > 1.5 for upregulation and < 0.66 for downregulation. These DEMs assigned to 21 metabolic pathways, according to KEGG analysis. Glycerophospholipid metabolism and sphingolipid metabolism represented the most important enriched metabolic pathways, combined with the analysis of the number of enriched DEMs and P-values. The top 10 upregulated and downregulated DEMs are listed in Table 3. Gossyrubilone and saxagliptin were the upregulated and downregulated DEMs with significant expression changes in the metabolomic profiling analysis of both androgenic gland and testis, respectively.

Principal component analysis (PCA) of metabolic profiles of the androgenic gland samples of M. nipponense at non-reproductive season and reproductive season

Orthogonal projections to latent structures discriminate analysis (OPLS-DA) analysis of the androgenic gland samples at non-reproductive season and reproductive season. OPLS-DA score plots based on the LC–MS spectra
Transcriptome profiling analysis
This study assembled 36,717 non-redundant transcripts with an average length of 1143.87 bp, and ranged from 301 bp to 29,020 bp. Approximately, 24.96% of non-redundant transcripts were 301–400 bp in length, followed by 401–500 bp (14.85%) and > 2000 bp (14.46%).
All of the assembled unigenes were then annotated in the Nr database in NCBI, GO, COG, and KEGG databases, in order to predict their putative functions, using an E-value of < 10−5. A total of 12,819 (34.91%) unigenes were annotated in the Nr database. The other unannotated unigenes represented the novel genes with functions that have not yet been clearly defined. A total of 9372 (25.52%) and 8856 (24.12%) were annotated in the GO and COG databases, respectively. GO analysis included three functional categories, which were biological process (22 functional groups), cellular component (17 functional groups), and molecular function (15 functional groups), of which cellular process, cell, cell part, and binding represent the main functional groups (Fig. 4). COG analysis identified 25 functional groups, which include general function prediction only, signal transduction mechanisms, secondary metabolite synthesis, transport and catabolism, and function unknown represent the main functional groups (Fig. 5). A total of 6787 (18.48%) unigenes matched the known proteins in the KEGG database and were mapped onto 217 metabolic pathways.

Gene ontology classification of non-redundant transcripts. The left y-axis indicates the percentage of a specific category of genes existed in the main category, whereas the right y-axis indicates the number of a specific category of genes existed in main category

Cluster of orthologous groups (COG) classification of putative proteins
A total of 9734 unigenes were differentially expressed in the androgenic gland between the reproductive vs. non-reproductive seasons using the criteria of > 1.5 for upregulation and < 0.66 for downregulation, of which 5256 unigenes were upregulated, and 4478 unigenes were downregulated during the reproductive season. These DEGs ranging from 1 to 54 were mapped onto 174 metabolic pathways. Phagosome, spliceosome, and ribosome biogenesis in eukaryotes, RNA degradation, and oxidative phosphorylation represented the main enriched metabolic pathways of DEGs, combined with the analysis of enriched DEGs and P-values.
Identification of reproduction-related genes
A total of 14 DEGs were selected from the main enriched metabolic pathways, based on the expression patterns noted in the samples from the non-reproductive vs. reproductive seasons, which were considered as strong candidate genes involved in male sexual development (Table 4). Proteasome alpha 3 and proteasome subunit beta type-5 were selected from the metabolic pathway of proteasome. Nucleolar protein 56 (NOL56), 5′-3′ exoribonuclease 1, and RNA exonuclease 1 homolog were selected from the metabolic pathway of ribosome biogenesis in eukaryotes. DEAD box polypeptide 39, Mago nashi 1, U2 snRNP-associated protein, and transcription elongation regulator 1 (TCERG1) were selected from the metabolic pathway of spliceosome. 6-phosphofructokinase, heat shock protein 60 (HSP60), and M-phase phosphoprotein 6 protein selected from the metabolic pathway of RNA degradation. Cytochrome oxidase subunit 3 and protein kinase C were selected from the metabolic pathway of oxidative phosphorylation. The expression of these 14 DEGs between the reproductive vs. non-reproductive seasons was further verified by qPCR analysis, which showed the same expression pattern as RNA-Seq (Fig. 6).

Verification of the expressions of 14 differentially expressed genes (DEGs) in the androgenic gland between non-reproductive season vs. reproductive season by qPCR. The amounts of DEGs expression were normalized to the EIF transcript level. Data are shown as mean ± SD (standard deviation) of tissues in three separate individuals. Lowercase indicates the expression difference of DEGs in the androgenic gland between non-reproductive season vs. reproductive season
qPCR analysis of DEGs in different tissues
The expression levels of these 14 DEGs were further verified in different mature tissues, in order to analyze their potential biological functions (Fig. 7). A total of 6 DEGs were highly expressed in the male sexual developmental system (testis and androgenic gland) in M. nipponense and showed significant differences with other tested tissues (p < 0.05), including NOL56, 5′-3′ exoribonuclease 1, RNA exonuclease 1 homolog, Mago nashi 1, U2 snRNP-associated protein, and TCERG1. Among these 6 DEGs, the expression levels of NOL56, 5′-3′ exoribonuclease 1, U2 snRNP-associated protein, and TCERG1 in testis showed significant difference with other tested tissues (p < 0.05), while RNA exonuclease 1 homolog and Mago nashi 1 showed significant differences in both the testis and androgenic gland compared with other tested tissues (p < 0.05). In addition, HSP60 and cytochrome oxidase subunit 3 also exhibited extremely high expression levels in male sexual developmental system.

Identification of the expressions of 14 DEGs in different tissues by qPCR. The amounts of DEGs expression were normalized to the EIF transcript level. Data are shown as mean ± SD (standard deviation) of tissues in three separate individuals. Lowercase indicate expression difference of DEGs in different tissues
Discussion
There were significant differences in the reproductive capacity between the M. nipponense prawns collected during the reproductive vs. non-reproductive seasons. Androgenic gland has been reported to play essential roles in promoting male sexual differentiation and development in crustaceans (Rosen et al. 2010; Ventura et al. 2009, 2011). It is widely acknowledged that environmental factors have considerable effects on the process of sexual differentiation and development in aquatic animals (Wedekind 2017). However, the regulatory effects of environmental factors on the reproductive capacity of the androgenic gland are still unclear. In this study, the morphological differences of the androgenic gland of prawns collected during the reproductive vs. non-reproductive seasons were determined by HE staining. DEMs and DEGs were selected from the androgenic gland of prawns from the two seasons which were examined via an integrated metabolome and transcriptome analysis.
In this study, the R2X(cum), R2Y(cum), and Q2(cum) were 0.942, 1, and 0.947, respectively. The values indicated the separation of the variation in the statistical data between the androgenic gland during the non-reproductive vs. reproductive seasons. The R2 and Q2 intercept values were 0.943 and − 0.906, respectively. This suggested a strongly robust and reliable model with a low risk of overfitting to identify the different metabolic patterns in the androgenic gland during the non-reproductive vs. reproductive seasons. A total of 260 DEMs were identified in this study. Glycerophospholipid metabolism and sphingolipid metabolism were the main enriched metabolic pathways of DEMs in the androgenic gland between the reproductive vs. non-reproductive seasons. A previous study also reported that glycerophospholipid metabolism and sphingolipid metabolism were the main enriched metabolic pathways of DEMs in the testis between the reproductive vs. non-reproductive seasons (Jin et al. 2020). Histological observations revealed significant morphological differences in the androgenic gland between the reproductive vs. non-reproductive seasons in this study. Thus, glycerophospholipid metabolism and sphingolipid metabolism were predicted to play essential roles in the development of the androgenic gland in the reproductive season, promoting the lipid accumulation during this time. Glycerophospholipids are the most common and abundant phospholipid in the body. In glycerophospholipids, two hydroxyl groups of glycerol bind with the fatty acids to form the esters, and the third hydroxyl group is esterified by phosphorylation to form the phosphatidylic acid (Dennis et al. 1991; Exton 1994; Meagher and Fitzgerald 1993). Glycerophospholipid plays essential roles in the formation of biofilms. In addition, glycerophospholipids are also important components of bile and membrane surfactant, as these participate in protein recognition and signal transduction in the cell membrane. Sphingolipids are amphoteric lipids, containing a sphingosine skeleton (Meagher and Fitzgerald 1993). Sphingolipids, including sphingomyelin and gangliosides, generally exist in the plant and animal membranes, especially in the central nervous system. The hydrolysis of sphingomyelin in cells is catalyzed by the nerve phospholipase in lysosomes. The hydrolysates are ceramide and choline phosphate, which can continue to metabolize (Spiegel and Merrill 1996). A congenital defect of phospholipase leads to the accumulation of sphingomyelin in tissues, leading to liver and spleen swelling, seriously affecting the central nervous system, and even resulting in death (Morales et al. 2007). Gossyrubilone and saxagliptin were the main DEMs in the metabolomic profiling analysis of both the androgenic gland and testis, which may play essential roles in the mechanism of male sexual development. Gossyrubilone is a dark red pigment, which is the isopentylimine of hemigossypolone and isolated from glands of young leaves of Gossypium (Bell et al. 1978). Saxagliptin was reported to be involved in the treatment of diabetes (Defronzo et al. 2009; Rosenstock et al. 2009).
In this study, a total of 36,717 non-redundant transcripts were assembled, providing valuable data for further studies of male sexual development in M. nipponense. Cellular process, cell, cell part, and binding were the main functional groups in GO analysis, and general function prediction only, signal transduction mechanisms, secondary metabolite synthesis, transport and catabolism, and function unknown were the main functional groups identified in COG analysis, which were consistent with previous studies on male sexual development in M. nipponense (Jin et al. 2020, 2021b, c). This indicates that these functional groups may consist of male sexual development-related genes, promoting further studies on male sexual development in M. nipponense. A total of 9734 unigenes were differentially expressed in the androgenic gland between the reproductive vs. non-reproductive seasons. Phagosome, spliceosome, ribosome biogenesis in eukaryotes, RNA degradation, and oxidative phosphorylation were the main enriched metabolic pathways of DEGs. Phagosome, spliceosome, and oxidative phosphorylation were also reported to be the main enriched metabolic pathways of DEGs in the testis between the reproductive vs. non-reproductive seasons (Jin et al. 2020). Glycerophospholipid metabolism and sphingolipid metabolism were also the main enriched metabolic pathways of DEMs in both the androgenic gland and testis between the reproductive vs. non-reproductive seasons. These results imply that the development of the androgenic gland has some regulatory relationship with testis development in M. nipponense, and this is consistent with previous reports which showed that the androgenic gland and its secreted hormones promote the development of male secondary characterization in crustacean species (Rosen et al. 2010; Ventura et al. 2009, 2011). The expression differences of 14 DEGs between the reproductive vs. non-reproductive seasons were verified by qPCR and showed the same expression pattern with those of RNA-Seq, indicating the accuracy of RNA-Seq.
Spliceosome and phagosome were the most enriched metabolic pathways, and RNA degradation was a main metabolic pathway in the transcriptome profiling analysis of the androgenic gland between the reproductive vs. non-reproductive seasons in M. nipponense. Spliceosome plays essential roles in the splicing of the coding regions of precursor messenger RNA (pre-mRNA). Spliceosome plays functions by combining distant regions of pre-mRNA with spliceosomal snRNAs and catalytic proteins (Will and Lührmann 2011). Phagocytosis plays essential roles in the tissue remodeling, inflammation, and defense against infectious agents. Phagocytosis is the process where large particles are absorbed by a cell (Fu and Tom 1990). A phagosome is formed through combination of the specific receptors on the phagocyte surface and ligands on the particle surface. Most bacteria will be killed and degraded into fragments by toxic products, released through the fusion of phagosomes and lysosomes (Hampton et al. 1998). RNA degradation plays essential roles in the degradation of redundant RNA in animals. It is widely acknowledged that cells transcribe more RNA than they accumulate. This implies that RNA degradation systems exist. RNA is degraded at the end of its useful life. This process is dramatically long for a ribosomal RNA, but very short when the introns are excised. The surveillance mechanisms are rapidly active to degrade the RNA molecules with defects in processing, folding, or assembly with proteins. RNA degradation must be carefully controlled to accurately recognize target RNAs, as it is ubiquitous in all cells (Houseley and Tollervey 2009; Lacava et al. 2005). A reasonable explanation for the main enrichment of DEGs in the immune-related metabolic pathways is that the development of the androgenic gland is more active in the reproductive season than that of non-reproductive season. Thus, the aged or redundant cells need to be digested, and the new cells need to be differentiated, in order to maintain normal gonad development. Many important DEGs were selected from the metabolic pathways of spliceosome, and RNA degradation, based on the gene annotation and fold changes of the gene expression, which may play essential roles in promoting the reproductive capacity in the reproductive season. U2 snRNP-associated protein and TCERG1 were vital DEGs from the metabolic pathway of spliceosome, which were upregulated in the reproductive season. The excision of introns from nuclear pre-mRNA requires assembly of the spliceosome from small nuclear ribonucleoprotein particles (snRNPs) (Guthrie 1991). A large number of the snRNPs have been identified to be required for pre-mRNA splicing in yeast through biochemical and molecular analyses (Beggs 1993; Ruby and Abelson 1991; Vijayraghavan et al. 1989). These snRNPs included U1, U2, U4, U5, and U6. The mutually exclusive U2-U6 snRNA interaction is necessary for catalytic activation. TCERG1 was reported to play essential roles in transcriptional elongation and alternative splicing of pre-mRNAs (Goldstrohm et al. 2001; Sánchez-Alvarez et al. 2006). Increasing evidence indicates that TCERG1 has potential roles in the coupling between transcription and splicing. The process of alternative pre-mRNA splicing of several genes was proven to be affected by TCERG1, including β-globin, β-tropomyosin, CD44, and fibronectin splicing reporters (Cheng et al. 2007; Lin et al. 2004; Pearson et al. 2008; Sánchez-Alvarez et al. 2010). TCERG1 also has regulatory effects on the alternative splicing of putative cellular targets, as seen through microarray analysis following TCERG1 knockdown (Pearson et al. 2008). TCERG1 regulates HIV-1 transcription by increasing the rate of RNAPII elongation through the phosphorylation of serine 2 (Coiras et al. 2013). 6-phosphofructokinase is a vital DEG from the metabolic pathway of RNA degradation, which was upregulated in the reproductive season. 6-phosphofructokinase plays essential roles in controlling glycolysis and respiration in plants (Dixon and ap Rees 1980; Kobr and Beevers 1971; Ruffner and Hawker 1977). Citrate inhibits the activities of phosphofructokinase and could be responsible for the decreased rate of glycolysis in different animals across the animal kingdom.
Ribosomes play vital roles in making proteins and are responsible for the ribosomal biogenesis of the production and correct assembly of four rRNAs and 80 ribosomal proteins in eukaryotes. Ribosomal biogenesis requires hundreds of factors, which were not present in the mature particles. Ribosome biogenesis is stalled, and cell growth is terminated even under optimal growth conditions, when these proteins are absent. NOL56, 5′-3′ exoribonuclease 1, and RNA exonuclease 1 homolog are the most important DEGs from the metabolic pathway of ribosome biogenesis in eukaryotes. Nucleolar proteins have a broad range of basic biological processes, including organ morphogenesis, growth, differentiation, homeostasis, and neoplasia. The biological functions of several nucleolar proteins have been identified. Rbm19 is a nucleolar protein, which is essential for the production of the 18S ribosomal RNA during ribosome biogenesis in C. tentans and C. elegans (Bjork et al. 2002; Jin et al. 2002; Saijou et al. 2004). The nucleolar protein 4-like gene is expressed in multiple organs in zebrafish embryos, implying that it plays important roles in the process of embryogenesis in zebrafish (Supriya et al. 2015). RNA exonuclease is an exonuclease of ribonucleic acid (RNA), which is an enzyme, playing essential roles in degrading RNA and removing nucleotides at the 5′ or 3′ end (Moser et al. 1998). 5′-3′ exoribonuclease 1 was reported to promote higher amounts of essentially full-length mRNAs. 5′-3′ exoribonuclease 1 lacking 5′-3′ exoribonuclease 1 in yeast cells showed poly(A) deficient and partially lacked the 5′ cap structure (Hsu and Stevens 1993). RNA exonuclease 1 homolog plays vital roles in the maintenance of stress response signaling and electrotaxis behavior in many animals (Taylor et al. 2021). In addition, in vitro culture revealed that the expression of RNA exonuclease homolog 1 transcript was primarily restricted in undifferentiated epiblasts (Blomberg et al. 2007).
Adenosine-triphosphate (ATP) is a high-energy compound, which is used as an energy source in nearly all metabolic activities, including male sexual development, especially that for the testis development. Many previous studies indicated that rapid gonad development has negative effects on the growth performance of aquatic species. Oxidative phosphorylation was the main enriched metabolic pathway in this study. Interestingly, a previous study also reported that oxidative phosphorylation was the main enriched metabolic pathway in the transcriptome profiling analysis of testis between the reproductive vs. non-reproductive seasons (Jin et al. 2020). Oxidative phosphorylation occurs in the cytoplasm of prokaryotes or in the inner membrane of the mitochondria of eukaryotic cells. The energy released from the oxidation of substances in vivo, which has positive effects on the coupling reaction between inorganic phosphate and adenosine diphosphate. ATP is synthesized through the respiratory chain (Dimroth et al. 2000). A reasonable explanation for this is that oxidative phosphorylation and glycolysis/gluconeogenesis provides energy for the sexual development of male M. nipponense during the reproductive season. Cytochrome oxidase subunit 3 and protein kinase C were selected from the metabolic pathway of oxidative phosphorylation. Cytochrome oxidase has significant regulatory roles in aerobic life, which blends the properties of several other metalloproteins, playing essential roles in the functions of either transport or redox. The preparations of cytochrome oxidase normally contain a variety of phospholipids, as well as some copper and iron (Lunt et al. 2019; Sunnucks and Hales 1996). Protein kinase C (PKC) is a family of serine/threonine protein kinases. PKCs have functions on their substrates at serine or threonine residues through phosphorylation. PKCs were found to play essential roles in controlling the function of other proteins and in several signal transduction cascades. PKC enzymes are activated by the increase of the concentration of DAG or calcium ions (Ca2+) (Good et al. 1998; Inoguchi et al. 2000).
The qPCR analysis in different mature tissues revealed that 6 out of 14 DEGs showed the highest expression levels in the testis and/or androgenic gland in M. nipponense. These 6 DEGs included NOL56, 5′-3′ exoribonuclease 1, RNA exonuclease 1 homolog, Mago nashi 1, U2 snRNP-associated protein, and TCERG1. Among these 6 DEGs, the expression levels of NOL56, 5′-3′ exoribonuclease 1, U2 snRNP-associated protein, and TCERG1 were highest in the testis, while RNA exonuclease 1 homolog and Mago nashi 1 showed high expression in both the testis and androgenic gland. This indicated that these 6 DEGs can be considered as strong candidate sex-related genes, which may be involved in the mechanism of male sexual differentiation and development in M. nipponense. In addition, HSP60 and cytochrome oxidase subunit 3 also showed dramatically high expression levels in the testis and androgenic gland, suggesting their potential roles in male sexual differentiation and development.
In conclusion, the present study identified the metabolites and genes which may play essential roles in sexual differentiation and development in male M. nipponense. This was done the integrated analysis of transcriptomic and metabolomic analysis of the androgenic gland between the reproductive vs. non-reproductive seasons. Glycerophospholipid metabolism and sphingolipid metabolism were the main metabolic pathways of DEMs, and phagosome, spliceosome, and oxidative phosphorylation represented the main enriched metabolic pathways of DEGs in this study, which was consistent with that of metabolomic analysis in the testis during the non-reproductive vs. reproductive seasons. This suggested that the androgenic gland plays a regulatory role in testis development. Eight genes were identified as strong candidate of sex-related genes, promoting the process of male sexual differentiation and development of M. nipponense, according to qPCR analysis in various mature tissues, which showed the highest expression levels in testis and/or androgenic gland. Overall, the important metabolites and genes were selected from the androgenic gland of M. nipponense, providing valuable evidences for further studies on the mechanism of male sexual development in M. nipponense and other crustacean species.
Data availability
The reads of M. nipponense transcriptome were submitted to NCBI with the accession number of SRX5805440-SRX5805445. The data of M. nipponense were submitted to MetaboLights with the accession number of MTBLS1025.
Code availability
Not applicable.
References
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. Nat Genet 25(1):25–29
Beggs JD (1993) Yeast protein factors involved in pre-mRNA splicing. Mol Biol Rep 18:99–103
Bell AA, Stipanovic RD, O’Brien DH, Fryxellc PA (1978) Sesquiterpenoid aldehyde quinones and derivatives in pigment glands of Gossypium. Phytochemistry 17(8):1297–1305
Benjamini Y, Drai D, Elmer G et al (2001) Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125(1–2):279–284
Bjork P, Bauren G, Jin S et al (2002) A novel conserved RNA-binding domain protein, RBD-1, is essential for ribosome biogenesis. Mol Biol Cell 13:3683–3695
Blomberg LA, Schreier LL, Talbot NC (2007) Expression analysis of pluripotency factors in the undifferentiated porcine inner cell mass and epiblast during in vitro culture. Mol Reprod Dev 75(3):450–463
Cheng D, Côté J, Shaaban S, Bedford MT (2007) The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell 25:71–83
Coiras M, Montes M, Montanuy I et al (2013) Transcription elongation regulator 1 (TCERG1) regulates competent RNA polymerase II-mediated elongation of HIV-1 transcription and facilitates efficient viral replication. Retrovirology 10:124
Defronzo RA, Hissa MN, Garber AJ et al (2009) The efficacy and safety of saxagliptin when added to metformin therapy in patients with inadequately controlled Type 2 diabetes with metformin alone. Diabetes Care 32(9):1649–1655
Dennis EA, Rhee SG, Billah MM, Hannun YA (1991) Role of phospholipase in generating lipid second messengers in signal transduction. FASEB J 5:2068–2077
Dimroth P, Kaim G, Matthey U (2000) Crucial role of the membrane potential for ATP synthesis by F(1)F(o) ATP synthases. J Exp Biol 203:51–59
Dixon WL, ap Rees T (1980) Identification of the regulatory steps in glycolysis in potato tubers. Phytochemistry 19:1297–1301
Exton JH (1994) Phosphatidylcholine breakdown and signal transduction. Biochim Biophys Acta 1212:26–42
Fu XD, Tom M (1990) Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature 343:437–441
Fu HT, Jiang SF, Xiong YW (2012) Current status and prospects of farming the giant river prawn (Macrobrachium rosenbergii) and the oriental river prawn (Macrobrachium nipponense) in china. Aquac Res 43:993–998
Goldstrohm AC, Greenleaf AL, Garcia-Blanco MA (2001) Co-transcriptional splicing of pre-messenger RNAs: considerations for the mechanism of alternative splicing. Gene 277:31–47
Good JA, Ziegler WH, Parekh DB et al (1998) Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 281(5385):2042–2045
Grabherr MG, Haas BJ, Yassour M et al (2011) Trinity: reconstructing a full-lengthtranscriptome without a genome from RNA-Seq data. Nat Biotechnol 29(7):644–652
Guthrie C (1991) Messenger RNA splicing in yeast: clues as to why the spliceosome is a ribonucleoprotein. Science 253:157–163
Hampton MB, Kettle AJ, Winterbourn CC (1998) Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92(9):3007–3017
Houseley J, Tollervey D (2009) The many pathways of RNA degradation. Cell 136(4):763–776
Hsu CL, Stevens A (1993) Yeast cells lacking 5’ 3’ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5’ cap structure. Mol Cell Biol 13:4826–4835
Hu YN, Fu HT, Qiao H et al (2018) Validation and evaluation of reference genes for Quantitative real-time PCR in Macrobrachium nipponense. Int J Mol Sci 19(8):2258
Huang XS, Ye HH, Huang HY et al (2014) An insulin-like androgenic gland hormone gene in the mud crab, Scylla paramamosain, extensively expressed and involved in the processes of growth and female reproduction. Gen Comp Endocrinol 204:229–238
Inoguchi T, Li P, Umeda F (2000) High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H Oxidase in Culutred Vascular Cells. Diabetes 49(11):1939–1945
Jin SB, Fu HT, Zhou Q et al (2013) Transcriptome analysis of androgenic gland for discovery of novel genes from the oriental river prawn, Macrobrachium nipponense, using Illumina Hiseq 2000. PLoS ONE 8:e76840
Jin SB, Fu HT, Sun SM et al (2018) iTRAQ-based quantitative proteomic analysis of the androgenic glands of the oriental river prawn, Macrobrachium nipponense, during nonreproductive and reproductive seasons. Comp Biochem Physiol Part D Genomics Proteomics 26:50–57
Jin SB, Hu YN, Fu HT et al (2019a) Potential functions of Gem-associated protein 2-like isoform X1 in the oriental river prawn Macrobrachium nipponense: cloning, qPCR, in situ hybridization, and RNAi analysis. Int J Mol Sci 20:3995
Jin SB, Hu YN, Fu HT et al (2019b) Identification of potentially novel functions of DNA polymerase zeta catalytic subunit in oriental river prawn, Macrobrachium nipoponense: Cloning, qPCR, in situ hybridization and RNAi analysis. 3Biotech 9:330
Jin SB, Hu YN, Fu HT et al (2020) Analysis of testis metabolome and transcriptome from the oriental river prawn (Macrobrachium nipponense) in response to different temperatures and illumination times. Comp Biochem Physiol Part D Genomics Proteomics 34:100662
Jin SB, Hu YN, Fu HT et al (2021a) Identification and characterization of the succinate dehydrogenase complex iron sulfur subunit B gene in the oriental river prawn Macrobrachium Nipponense. Front Genet 12:698318
Jin SB, Fu Y, Hu YN et al (2021b) Identification of candidate genes from androgenic gland in Macrobrachium nipponense regulated by eyestalk ablation. Sci Rep 11:1985
Jin SB, Fu Y, Hu YN et al (2021c) Transcriptome profiling analysis of the testis after eyestalk ablation for selection of the candidate genes involved in the male sexual development in Macrobrachium nipponense. Front Genet 12:675928
Jin SB, Zhao J, Bjork P et al (2002) Mrd1p is required for processing of pre-rRNA and for maintenance of steady-state levels of 40 S ribosomal subunits in yeast. J Biol Chem 277:18431–18439
Kanehisa M, Araki M, Goto S et al (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:480–484
Kobr MJ, Beevers H (1971) Gluconeogenesis in the castor bean endosperm. Plant Physiol 47:48–52
Lacava J, Houseley J, Saveanu C et al (2005) RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121(5):713–724
Leslie LH, Valentine AA (2015) 5-DMRT1 and the road to masculinity. Sertoli Cell Biology. Second edn. Academic Press pp 123–174
Li SH, Li FH, Sun Z, Xiang JH (2012) Two spliced variants of insulin-like androgenic gland hormone gene in the Chinese shrimp Fenneropenaeus Chinensis. Gen Comp Endocrinol 177(2):246–255
Lin KT, Lu RM, Tarn WY (2004) The WW domain-containing proteins interact with the early spliceosome and participate in pre-mRNA splicing in vivo. Mol Cell Biol 24:9176–9185
Liu F, Shi W, Ye H et al (2021) RNAi reveals role of insulin-like androgenic gland hormone 2 (IAG2) in sexual differentiation and growth in Hermaphrodite Shrimp. Front Mar Sci 8:666763
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 25:402–408
Lunt DH, Zhang DX, Szymura JM, Hewitt GM (2019) The insect cytochrome oxidase I gene: evolutionary patterns and conserved primers for phylogenetic studies. Insect Mol Biol 5(3):153–165
Ma XK, Liu XZ, Wen HS et al (2006) Histological observation on gonadal sex differentiation in Cynoglossus semilaevis Günther. Mar Fish Res 27(2):55–61
Ma KY, Li JL, Qiu GF (2016) Identification of putative regulatory region of insulin-like androgenic gland hormone gene (IAG) in the prawn Macrobrachium nipponense and proteins that interact with IAG by using yeast two-hybrid system. Gen Comp Endocrinol 229:112–118
Meagher EA, Fitzgerald GA (1993) The molecular basis of eicosanoid action. Hypertens Pregnancy 12:439–451
Morales A, Lee H, Goñi FM et al (2007) Sphingolipids and cell death. Apoptosis 12(5):923–939
Moser MJ, Holley WR, Chatterjee A, Mian IS (1998) The proofreading domain of Escherichia coli DNA polymerase I and other DNA and/or RNA exonuclease domains. Nucleic Acids Res 25(24):5110–5118
Om AD, Sharif S, Jasmani S, Sung YY, Bolong AA (2015) Molecular characteristic ofgiant grouper (Epinephelus Lanceolatus) Vitellogenin. J Aquac Res Development 6(9):1000360
Ortiz-Villanueva E, Navarro-Martín L, Jaumot J, et al (2017) Metabolic disruption of zebrafish (Danio rerio) embryos by bisphenol A. An integrated metabolomic and transcriptomic approach. Environ Pollut 231(Pt1): 22–36
Pearson JL, Robinson TJ, Muñoz MJ et al (2008) Identification of the cellular targets of the transcription factor TCERG1 reveals a prevalent role in mRNA processing. J Biol Chem 283:7949–7961
Rosen O, Manor R, Weil S et al (2010) A sexual shift induced by silencing of a single insulin-like gene in crayfish: ovarian upregulation and testicular degeneration. PLoS ONE 5:e15281
Rosenstock J, Aguilar-Salinas C, Klein E et al (2009) Effect of saxagliptin monotherapy in treatment-naïve patients with type 2 diabetes. Curr Med Res Opin 25(10):2401–2411
Ruby SW, Abelson J (1991) Pre-mRNA splicing in yeast. Trends Genet 7:79–85
Ruffner HP, Hawker JS (1977) Control of glycolysis in Vitis vinifera. Phytochemistry 16:1171–1175
Sagi A, Cohen D, Wax Y (1986) Production of Macrobrachium rosenbetgii in momosex population: yield characterises under intensive monoculture conditions in cages. Aquaculture 51(3–4):265–275
Sagi A, Cohen D, Milner Y (1990) Effect of androgenic gland ablation on morphotypic differentiation and sexual characteristics of male freshwater prawns, Macrobrachium rosenbergii. Gen Comp Endocr 77:15–22
Saijou E, Fujiwara T, Suzaki T et al (2004) RBD-1, a nucleolar RNA-binding protein, is essential for Caenorhabditis elegans early development through 18S ribosomal RNA processing. Nucleic Acids Res 32:1028–1036
Sánchez-Alvarez M, Goldstrohm AC, Garcia-Blanco MA, Suñé C (2006) Human transcription elongation factor CA150 localizes to splicing factor-rich nuclear speckles and assembles transcription and splicing components into complexes through its amino and carboxyl regions. Mol Cell Biol 26:4998–5014
Sánchez-Alvarez M, Montes M, Sánchez-Hernández N et al (2010) Differential effects of sumoylation on transcription and alternative splicing by transcription elongation regulator 1 (TCERG1). J Biol Chem 285:15220–15233
ShangGuan BM, Liu ZZ, Li SQ (1991) Histological studies on ovarian development in Scylla serrata. J Fish China 15(2):96–103
Spiegel S, Merrill AH (1996) Sphingolipid metabolism 622 and cell growth regulation. FASEB J 10(12):1388–1397
Sunnucks P, Hales DF (1996) Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol Biol Evol 13(3):510–524
Supriya B, Praveen B, Rajeeb S (2015) Nucleolar protein 4-like has a complex expression pattern in zebrafish embryos. Int J Dev Biol 52:1–3
Tatusov RL, Fedorova ND, Jackson JD et al (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4(1):41
Taylor SKB, Minhas MH, Tong J et al (2021) C. elegans electrotaxis behavior is modulated by heat shock response and unfolded protein response signaling pathways. Sci Rep 11:3115
Ventura T, Manor R, Aflalo ED et al (2009) Temporal silencing of an androgenic gland-specific insulin-like gene affecting phenotypical gender differences and spermatogenesis. Endocrinology 150:1278–1286
Ventura T, Manor R, Aflalo ED et al (2011) Expression of an androgenic gland-specific insulin-like peptide during the course of prawn sexual and morphotypic differentiation. ISRN Endocrinol 2011:476283
Ventura T, Manor R, Aflalo ED et al (2012) Timing sexual differentiation: full functional sex reversal achieved through silencing of a single insulin-like gene in the prawn Macrobrachium Rosenbergii. Biol Reprod 86:90
Vijayraghavan U, Company M, Abelson J (1989) Isolation and characterization of pre-mRNA splicing mutants of S. cerevisiae. Genes Dev 3:1206–1216
Wedekind C (2017) Demographic and genetic consequences of disturbed sex determination. Philos Trans R Soc Lond B Biol Sci 372(1729):20160326
Will CL, Lührmann R (2011) Spliceosome structure and function. Csh Perspect Biol 3(7):21441581
Zhang YP, Fu HT, Qiao H et al (2013) Molecular cloning and expression analysis of transformer-2 gene during development in Macrobrachium nipponense (de Haan 1849). J World Aquacult Soc 44(3):338–349
Zhang XL, Cui LF, Li SM et al (2020) Bureau of Fisheries, Ministry of Agriculture, P.R.C. Fisheries economic statistics. In: China Fishery Yearbook. China Fishery Statistical Yearbook. Beijing China Agricultural Press 24.
Zhou TT, Wang W, Wang CG et al (2021) Insulin-like androgenic gland hormone from the shrimp Fenneropenaeus merguiensis: expression, gene organization and transcript variants. Gene 782:145529
Funding
This research was supported by grants from the National Key R&D Program of China (2018YFD0900201); Central Public-interest Scientific Institution Basal Research Fund CAFS (2021JBFM02; 2020TD36); Jiangsu Agricultural Industry Technology System; the China Agriculture Research System-48 (CARS-48); the New cultivar breeding Major Project of Jiangsu province (PZCZ201745). Thank you for the Jiangsu Province Platform for the Conservation and Utilization of Agricultural Germplasm.
Author information
Authors and Affiliations
Contributions
S.J. designed and wrote the manuscript. W.Z. performed the qPCR analysis. S.J. and Y.X. provided the experimental prawns. H.Q. selected the differentially expressed metabolites. Y.G. selected the differentially expressed genes. Y.W. performed the histological observations. H.F. supervised the experiment.
Corresponding author
Ethics declarations
Ethics approval
Permission was obtained from the Tai Lake Fishery Management Council and the committee of Freshwater Fisheries Research Center during the experimental programs. All experiments were performed in accordance with relevant guidelines and regulations.
Conflict of interest
The authors declare no competing interests.
Additional information
Handling Editor: Gavin Burnell
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jin, S., Zhang, W., Xiong, Y. et al. Genetic regulation of male sexual development in the oriental river prawn Macrobrachium nipponense during reproductive vs. non-reproductive season. Aquacult Int 30, 2059–2079 (2022). https://doi.org/10.1007/s10499-022-00887-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10499-022-00887-7