
Abstract
To select a reliable and sensitive method for discriminating strains of Porphyra haitanensis, the nucleotide sequence of the internal transcribed spacer 1 to internal transcribed spacer 2 regions (ITS-5.8S) of nuclear ribosomal DNA and the intergenic spacer region of RUBISCO were compared in five wild and five cultivated Porphyra haitanensis strains. Based on molecular analyses, sequences of ITS-5.8S (about 1,210 bp) could be divided into three regions: ITS1, 5.8S, and ITS2. The ITS1 and ITS2 sequences of each strain differed, even between individuals collected from the same site. In contrast, 5.8S rDNA and RUBISCO spacer sequences were identical among the ten P. haitanensis strains, although differences were found among different Porphyra species. Phylogenetic analysis also supported these conclusions. These sequence features of highly conserved regions and diversified regions that occurred repeatedly in ITS-5.8S could be useful in discriminating germplasm of P. haitanensis strains or Porphyra species. In contrast, the RUBISCO spacer is only suitable for identifying Porphyra species. New coupled primers were designed to amplify only the 5.8S rDNA and ITS2 region of Porphyra. The sequences of these amplified fragments can be readily used to identify germplasm or to perform phylogenetic analysis of Porphyra spp.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Porphyra, a genus of marine red algae, is an important economic marine crop, with an annual harvest of more than 130,000 t (dry weight) and a value of over US$2 billion. Today, the farming and processing of Porphyra have generated large seaweed industries in East Asian countries, including China, Japan, South Korea, and North Korea (Sahoo et al. 2002). Among Porphyra species, P. haitanensis is one of the most economically important. It has been widely cultivated along the coasts of South China, especially in the Fujian and Zhejiang Provinces.
In recent years, the production of P. haitanensis has constituted 75% of the total production of cultivated Porphyra in China (Zhang et al. 2005). With the advances of biotechnology and Porphyra farming, Porphyra cultivators are now carrying out intensive selections of P. haitanensis breed varieties and have selected many strains for cultivation in South China. For any cultivar, the correct identification of species or forma of the cultivated strains is necessary to ensure a well-bred cultivation and good production quality. However, because the gametophytic blade of Porphyra is morphologically simple and marked variations can occur as environmental conditions change, it is very difficult to precisely identify the species or forma of cultivated strains based only on their morphological characteristics. In addition, germplasm material has often been maintained as free-living conchocelis in laboratory subcultures. These conchocelis strains often cannot be definitively identified even using detailed morphological observations.
With new advances in molecular biology, molecular markers and DNA fingerprinting techniques have become routine for the identification and classification of many crops, including seaweeds. In cultivated Porphyra, the discrimination of strains has been attempted with restriction fragment length polymorphism (RFLP) analysis (Mizukami et al. 1996), random amplified polymorphic DNA (RAPD) analysis (Jia et al. 2000), amplified fragment length polymorphism (AFLP) analysis (Niwa et al. 2004; Sun et al. 2005), sequence-related amplified polymorphism (SRAP) analysis (Qiao et al. 2007; Xie et al. 2008), and simple sequence repeats (SSRs) analysis (Liu et al. 2005; Sun et al. 2006a). However, the limitations of these markers, including poor consistency, low reproducibility, labor intensiveness, or high cost, also limit their utilization to a significant degree.
Recently, nucleotide sequence data for the nuclear small subunit (SSU) rRNA gene, the ribosomal internal transcribed spacer (ITS), the plastid RUBISCO large subunit (rbcL), and the rbcL-rbcS intergenic spacer (RUBISCO spacer) have been used to determine the interspecific or intraspecific relationships of various Porphyra species (Freshwater et al. 1994; Ragan et al. 1994; Oliveira et al. 1994; Brodie et al. 1998; Woolcott and King 1998; Kunimoto et al. 1999a, b, 2003; Lindstrom and Fredericq 2003; Niwa and Aruga 2003, 2005a, b, 2008a, b; Xie et al. 2007). The superiority of these approaches is that they are based on popular and well-established sequencing technologies and can provide definitive results of nucleotide sequences (Kouduka et al. 2007).
In Porphyra species, Kunimoto et al. (2003) have suggested that the exon sequence of the SSU rRNA gene is an appropriately defined genetic marker for characterizing Porphyra species and that the Porphyra intron sequences of the SSU rRNA gene and ITS1 are appropriate for cultivar identification. However, because of its complicated structure (including varied numbers of exons and introns) and its long sequence (Kunimoto et al. 1999a), the SSU rRNA gene is difficult to amplify by PCR and to sequence. Lindstrom and Fredericq (2003) reported that the rbcL gene has a better resolution than the SSU rRNA gene for elucidating species-level relationships in Porphyra species. However, the rbcL gene is inappropriate for elucidating intraspecific relationships in Porphyra species.
The nucleotide sequence of the ITS1 region and that of the RUBISCO spacer are assumed to have had substitutions many times more frequently than the rDNA exon region (Kunimoto et al. 1999b; Lindstrom and Fredericq 2003). Therefore, in present study, we have compared the nucleotide sequence of nuclear ribosomal DNA of the internal transcribed spacer 1 to internal transcribed spacer 2 region (ITS-5.8S) and of the plastid RUBISCO intergenic spacer region (RUBISCO spacer) for ten P. haitanensis strains. Our goal was to determine a reliable and sensitive method for discriminating strains of P. haitanensis.
Materials and methods
Materials
The free-living conchocelis (sporophyte phase of Porphyra) of ten strains of P. haitanensis were used in this study (Table 1). The cultivated strains of Q-1, Z-17, Z-26, and Z-61 of P. haitanensis have been selected and purified by the laboratory of germplasm improvements and applications in Jimei University, and all have been cultivated in a large scale on the coast of Fujian Province, China. Each strain has its own characteristics: Q-1 has high quality gametophytic blades, Z-17 grows rapidly, Z-26 can resist high temperatures, and the thallus of Z-61 is thick. The control cultivated strain (PXI) was a traditional cultivated strain from the coast of Fujian Province, China. The five wild strains were selected and purified from wild P. haitanensis populations off the coast of Fujian Province, China, and have been used as germplasm material in P. haitanensis breeding. All of the samples were stored in the laboratory of germplasm improvements and applications of P. haitanensis in Jimei University.
Stock cultures of the free-living conchocelis were maintained in the laboratory at 21 ± 1°C under 50–60 μmol photons m−2 s−1 (12L: 12D) provided by cool white fluorescent lamps. To obtain sufficient conchocelis clumps for DNA extraction, free-living conchocelis colonies were chopped into short fragments with a homogenizer and the conchocelis fragments were cultured in PES medium (Wang et al. 1986).
DNA extraction, amplification, and sequence analysis
DNA was isolated from the free-living conchocelis of each strain. The collected free-living conchocelis were ground into powder with high-speed homogenization, and DNA was extracted and purified by the method of CTAB (http://www.cimmyt.org/english/docs/manual/protocols/abc_amgl.pdf). DNA concentrations were determined with a DU-600 spectrophotometer (Beckman Coulter, Fullerton, CA, USA) and adjusted to 5 ng/μl for PCR amplification.
The regions of ITS-5.8S and RUBISCO spacer were amplified with the primers in Table 2.
For amplification of the two DNA fragments, the PCR mixture (25 μl) contained 2.5 μl 10× PCR Buffer,10 ng template DNA,2.5 mM Mg2+,1.5U Taq enzyme (Takara Biotechnology, Co., Ltd., Dalian, China),200 nM of each primer (Takara),200 μM dNTP (Takara), and sterile water. The amplifications were performed in a MT programmable thermal controller PTC-200 (MJ. Research, Inc.). The PCR program was 95°C for 5 min, followed by 30 cycles of 94°C for 45 s, 55°C for 1 min, and 72°C for 2 min, and finally 72°C for 10 min. To confirm the presence of amplified DNA fragments, PCR products were loaded onto 1.5% agarose gels and stained with ethidium bromide. PCR products were gel purified with a Takara agarose gel DNA purification kit (Takara) following the manufacturer’s instructions. Sequencing was performed by Takara Biotechnology.
All of the novel sequence data were submitted to the GenBank. The accession numbers of ITS-5.8S and RUBISCO spacer are shown in Table 1.
Sequence identification, alignment, and phylogenetic analysis
Sequences were identified with Porphyra sequences existing in GenBank, using BLAST to confirm the boundary of each region.
The sequences determined in the present study and the additional corresponding sequences of other Porphyra species or strains, which were downloaded from GenBank (Table 3), were used in phylogenetic and genealogical analyses. The sequences were aligned using clustal X 1.83 (Thompson et al. 1997), and then refined by eyed. When all sequences had aligned, the base type that was in majority of the ten sequences seemed to be the intrinsic base in the variance positions. Phylogenetic relationships among these sequences were inferred using the neighbor-joining (NJ) method (Saitou and Nei 1987). The NJ method was performed with the MEGA4 (Tamura et al. 2007), and stability of the resulting groups was tested by bootstrap analysis over 1,000 replications.
Results
PCR amplification
Figures 1 and 2 show the PCR products of the ITS-5.8S and RUBISCO spacer, respectively, in ten P. haitanensis strains. A single band of amplified DNA was observed for each of the ten strains. The bands from the ten strains were of a similar size, at approximately 1.2 (ITS-5.8S) and 0.5 (RUBISCO spacer) kilobase pairs.

PCR profile of the ITS-5.8S for ten Porphyra haitanensis strains. Lane 1 Q-1, lane 2 z-17, lane 3 Z-26, lane 4 Z-61, lane 5 PXI, lane 6 DD1, lane 7 DD2, lane 8 DS, lane 9 JJS1, lane 10 JJS2, M shows molecular weight markers

PCR profile of the RUBISCO spacer for ten Porphyra haitanensis strains. Lane 1 Q-1, lane 2 z-17, lane 3 Z-26, lane 4 Z-61, lane 5 PXI, lane 6 DD1, lane 7 DD2, lane 8 DS, lane 9 JJS1, lane 10 JJS2, M shows the molecular weight markers
Nucleotide sequences and phylogenetic analyses of the ITS-5.8S
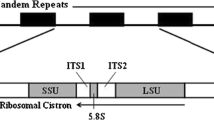
By multiple sequence alignments with existing Porphyra sequences in GenBank using BLAST, the structure and boundary of each part of ITS-5.8S of P. haitanensis could be defined. The two flanks of 5.8S had polyA and polyT sequences. Table 4 shows the details of ITS-5.8S of the ten P. haitanensis strains. The sequence length of ITS-5.8S ranged from 1,212 bps (PXI, DD1) to 1,221 bps (Q-1), and the base component of each sequence varied slightly. The 5.8S rDNA region of the ten strains was of an identical 160 bp size. The ITS1 and ITS2 region varied slightly by two or three bps between strains, due to the presence of indels, but were approximately 368 and 690 bp, respectively.
Table 5 shows the variable nucleotide positions of the ITS1 regions of the ten P. haitanensis strains by multiple sequence alignment. The sequence differences between the ten strains are shown in Table 6. The ITS1 sequences were all different from each other, and identical sequence pairs were not found among these strains, even between the strains that had been collected from the same site. Similar ITS1 sequences were found between Q-1 and Z-17, Z-17 and Z-61, Z-17 and JJS1, PXI and DS, PXI and JJS2, JJS1 and JJS2, which showed only one point change in 369 bp aligned sites. The maximum difference in ITS1 sequences were found between DD1 and Q-1, Z-26, and Z-61, which showed 7 points of variance in 369 aligned sites. On the ten P. haitanensis strains, the ITS1 sequence divergence (%) was between 0.27 and 1.63, with an average divergence of 1.06.
Table 7 shows the variable nucleotide positions of the ITS2 region of the ten P. haitanensis strains by multiple sequence alignment. The sequence differences between the ten strains are shown in Table 8. The ITS2 sequences were also different from each other, and identical sequence pairs were not found among these ten strains. Similar ITS2 sequences were found between Q-1 and Z-17, Z-17 and Z-61, Q-1 and Z-61, which showed only 2 points of variance in 696 bp aligned sites. The maximum difference of the ITS2 sequences were found between DS and Z-26, which showed 21 points of variance in 696 aligned sites. For the ten P. haitanensis strains, the ITS2 sequence divergence (%) was between 0.29 and 3.03, with an average divergence of 1.66. The average % sequence divergence between the wild strains was 1.80, which was higher than that between the cultured strains at 0.36.
A phylogenetic tree (Fig. 3) was produced by neighbor-joining (NJ) analysis, based on the ITS2 sequence dataset of the ten P. haitanensis strains, using MEGA 4.0. In this tree, the ten P. haitanensis strains could be separated into three groups—one group that included the four cultivated strains and the second group that was composed of the five wild strains. The control cultivated strain (PXI) formed another clade.

Cladogram based on ITS2 sequences, showing the relationship between the ten strains of Porphyra haitanensis. The tree was constructed using the NJ method and bootstrap value calculation from 1,000 trees
Identical 5.8S rDNA region sequences were found for the ten P. haitanensis strains, but different sequences of 5.8S were found among different Porphyra species (Table 9). The divergence of eight species of Porphyra was between 0 and 14.1%. P. drewiana and P. spiralis had identical sequences in their 5.8S rDNA regions. The phylogenetic tree (Fig. 4) that was produced by NJ analysis based on the 5.8S rDNA sequence dataset indicated a clear separation of the Porphyra species, except for P. drewiana and P. spiralis.

Cladogram based on 5.8S sequences, showing the relationship between eight species of Porphyra. The tree was constructed by using the NJ method and bootstrap value calculation from 1,000 trees
Nucleotide sequences and phylogenetic analyses of the RUBISCO spacer region
Similar to the 5.8S rDNA region, identical RUBISCO spacer sequences were also found for the ten P. haitanensis strains, while different Porphyra species had different sequences for their RUBISCO spacers (Table 10). The divergence of six of the species of Porphyra was between 1.9 and 9.3%, and no identical sequences were found between the Porphyra species. The phylogenetic tree (Fig. 5) that was produced by NJ analysis based on the rbcS-RUBISCO spacer sequence dataset also showed a clear separation of the Porphyra species.

Cladogram based on RUBISCO spacer sequences, showing the relationship between six species of Porphyra. The tree was constructed by using the NJ method and bootstrap value calculation from 1,000 trees
Discussion
In this study, we have addressed problems of strain identification of P. haitanensis using DNA sequencing of a small PCR product. Numerous papers have proposed that Porphyra strains of the same species can be discriminated by comparing the sequences of ITS1 regions (Kunimoto et al. 1999a, b, 2003; Niwa and Aruga 2003, 2005a, b, 2008a, b). Of the ten strains of P. haitanensis studied here, the sequences of the ITS-5.8S (about 1,210 bp) could be divided into three regions: ITS1, 5.8S and ITS2. The ITS sequence (including ITS1 and ITS2) of each strain was found to be different, and even differed between individuals collected from the same site. These sequences did tend to be very similar among all specimens, with variation scores < 3.03%, as listed in Tables 6 and 8. This result is in accord with several previous published studies (Kunimoto et al. 1999a, b, 2003; Niwa and Aruga 2003, 2005a, b, 2008a, b). The molecular features reported here will be of practical use for discrimination and establishing the genetic relationships of Porphyra strains.
The sequence divergence (%) of the ITS1 region between strains was not similar to that seen for the ITS2 region, even between the same strains (Tables 6, 8). This difference was unpredictable. The ITS1 sequence length is about 368 bp, and only one or two base differences were noted between the ten P. haitanensis strains (Table 4). The sequence divergence (%) was between 0.27 and 1.63 (average was 1.06), and in many strains, only one base substitution or gap difference was noted. The divergence among cultivated strains had less obvious differences than did the wild strains (Table 6).
For the ITS2, with a sequence length between 685 and 692 bp, the difference between strains was much greater than that seen for the ITS1. Sequence divergence (%) was between 0.29 and 3.03 (average was 1.66), which was also much higher than that of the ITS1 region. The divergence of ITS2 sequences among wild strains was clearly higher than that for cultivated strains (Table 8). Phylogenetic analysis based on the nucleotide sequences of ITS2 (Fig. 3) could clearly distinguish the wild and cultivated strains of P. haitanensis. The ITS2 region may, therefore, be more suitable than ITS1 for discriminating and establishing genetic relationships in P. haitanensis strains, as it offers a larger number of variable sites.
The 5.8S rDNA sequences were identical among the ten P. haitanensis strains. However, after blast analysis in GenBank, different sequences were found among different Porphyra species, except for P. drewiana and P. spiralis. This shows that the sequence of 5.8S differed among Porphyra species, while sequences from strains of the same species were identical. This sequence feature is similar to that seen for the SSU rDNA exon (Kunimoto et al. 1999a, b, 2003; Jones et al. 2004) and indicates that the nucleotide sequence of 5.8S RNA gene also can be used to discriminate between Porphyra species. The phylogenetic analysis clearly supported this conclusion (Fig. 4). The 5.8S RNA gene is only 160 bp lengths and has no introns, which makes it a much shorter and simpler gene than the SSU rDNA. We believe that the 5.8S RNA gene may, therefore, have a number of advantages over the SSU rDNA for the discrimination of Porphyra species.
Based on these results, one new set of coupled primers can be designed to amplify only the 5.8S rDNA and the ITS2 region of Porphyra. Sequences of the amplified fragments can then be easily used for germplasm identification and phylogenetic analysis of Porphyra. With the assistance of the Primer premier™ 5.0 and oligo 6.0 software, the new couple primers 5′-TTTGTCTTATCCGCTTTGT-3′ and 5′-GTGGTCCTACCTGATTTGAG-3′ were designed for this study. Their Tm value was 54°C and the 936 bp amplified fragment included the 5.8S rDNA and the ITS2 region. The results of PCR amplification (Fig. 6) and sequence data of PCR products using the new couple primers showed that the new couple primers work reliably.

PCR profile of the 5.8S-ITS2 for ten Porphyra haitanensis strains. Lane 1 Q-1, lane 2 z-17, lane 3 Z-26, lane 4 Z-61, lane 5 PXI, lane 6 DD1, lane 7 DD2, lane 8 DS, lane 9 JJS1, lane 10 JJS2, M shows molecular weight markers
The result of multi-sequence alignment of the RUBISCO spacer was similar to that seen for the 5.8S rDNA Identical sequences were also found for the ten P. haitanensis strains, but different Porphyra species had different sequences (Table 10). The same result has also been reported for P. yezoensis strains (Niwa et al. 2008a). Therefore, the RUBISCO spacer is not suitable for the discrimination of P. haitanensis strains, but it is a valuable marker for discriminating different Porphyra species (Brodie et al. 1998). The variation values of 5.8S rDNA and RUBISCO spacer, listed in Tables 9 and 10, are not distinctly different, so we cannot judge a priori which gene (5.8S rDNA or RUBISCO spacer) would be better suited for species identification in Porphyra.
From the above results, we can conclude that contrasting the nucleotide sequences of the 5.8S rDNA and the ITS2 regions would provide a reliable and sensitive method to discriminate Porphyra species or P. haitanensis strains. In contrast, the RUBISCO spacer is only suitable for identifying Porphyra species.
Abbreviations
- ITS-5.8S:
-
Nuclear ribosomal DNA of internal transcribed spacer 1 to internal transcribed spacer 2 region
- RUBISCO spacer:
-
Intergenic spacer region of RUBISCO
- RFLP:
-
Restriction fragment length polymorphism
- RAPD:
-
Random amplified polymorphic DNA
- AFLP:
-
Amplified fragment length polymorphism
- SRAP:
-
Sequence-related amplified polymorphism
- SSRs:
-
Simple sequence repeats
- SSU:
-
Nuclear small subunit
- rbcL:
-
Plastid RUBISCO large subunit
References
Brodie J, Hayes PK, Barker GL et al (1998) A reappraisal of Porphyra and Bangia (Bangiophycidae, Rhodophyta) in the northeast Atlantic based on the rbcL-rbcS intergenic spacer. J Phycol 34:1069–1074
Broom JE, Nelson WA, Yarish C et al (2002) A reassessment of the taxonomic status of Porphyra suborbiculata Kjellm, Porphyra carolinensis Coll et J. Cox and Porphyra lilliputiana W.A. Nelson, G.A. Knight et M.W. Hawkes (Bangiales, Rhodophyta) based on molecular and morphological data. Eur J Phycol 37:227–236
Freshwater DW, Fredericq S, Butler BS et al (1994) A gene phylogeny of the red algae (Rhodophyta) based on plastid rbcL. Proc Natl Acad Sci 91:7281–7285
Goff LJ, Moon DA (1993) PCR amplification of nuclear and plastid genes from algal herbarium specimens and algal spores. J Phycol 29:381–384
Hu ZM, He YJ, Duan DL (2007) Molecular identification of Chinese cultivated Porphyra (Bangiaceae, Rhodophyta) based on the rDNA internal transcribed spacer-1 sequence and random amplified polymorphic DNA markers. Mar Biol Res 3(1):20–28
Jia JH, Wang P, Jin DM et al (2000) The application of RAPD markers in diversity detection and variety identification of Porphyra. Acta Bot Sin 42(4):403–407
Jones WA, Griffin NJ, Jones DT et al (2004) Phylogenetic diversity in South African Porphyra (Bangiales, Rhodophyta) determined by nuclear SSU sequence analyses. EUR J Phycol 39:197–211
Kouduka M, Sato D, Komori M et al. (2007) A solution for universal classification of species based on genomic DNA. Int J Plant Genomics 27894
Kunimoto M, Kito H, Yamamoto Y et al (1999a) Discrimination of Porphyra species based on small subunit ribosomal RNA gene sequence. J Appl Phycol 11:203–209
Kunimoto M, Kito H, Kaminishi Y et al (1999b) Molecular divergence of the SSU rRNA gene and internal transcribed spacer 1 in Porphyra yezoensis (Rhodophyta). J Appl Phycol 11:211–216
Kunimoto M, Kito H, Mizukami Y et al (2003) Molecular features of a defined marker for the determination of the Porphyra tenera lineage. J Appl Phycol 15:337–343
Lindstrom SC, Fredericq S (2003) rbcL gene sequences reveal relationships among north-east Pacific species of Porphyra (Bangiales, Rhodophyta) and a new species, P. aestivalis. Phycol Res 51:211–224
Liu BQ, Zeng QG, Luo QJ et al (2005) Establishment of the DNA fingerprint of Porphyra haitanensis filament lines with microsatellite markers. J Fish Chin 29:323–326
Milstein D, Oliveira MC (2005) Molecular phylogeny of Bangiales (Rhodophyta) based on small subunit rDNA sequencing: emphasis on Brazilian Porphyra species. Phycologia 44(2):212–221
Mizukami Y, Okauti M, Kito H et al (1996) DNA fingerprinting of cultivated laver Porphyra yezoensis and P. tenera with oligonucleotide probes, and its application to cultivar discrimination. Fish Sci 62:173–177
Niwa K, Aruga Y (2003) Rapid DNA extraction from conchocelis and ITS-1 rDNA sequences of seven strains of cultivated Porphyra yezoensis (Bangiales, Rhodophyta). J Appl Phycol 15:29–35
Niwa K, Kikuchi N, Lwabuchi M et al (2004) Morphological and AFLP variation of Porphyra yezoensis Ueda form, narawaensis Miura (Bangiales, Rhodophyta). Phycol Res 52:180–190
Niwa K, Kikuchi N, Aruga Y (2005a) Morphological and molecular analysis of the endangered species Porphyra tenera (Bangiales, Rhodophyta). J Phycol 41:294–304
Niwa K, Kobiyama A, Aruga Y (2005b) Confirmation of cultivated Porphyra tenera (Bangiales, Rhodophyta) by polymerase chain reaction restriction fragment length polymorphism analyses of the plastid and nuclear DNA. Phycol Res 53:296–302
Niwa K, Kato A, Kobiyama A et al (2008a) Comparative study of wild and cultivated Porphyra yezoensis (Bangiales, Rhodophyta) based on molecular and morphological data. J Appl Phycol 20:261–270
Niwa K, Furuita H, Yamamoto T et al (2008b) Identification and characterization of a geeen-type mutant of Porphyra tenera Kjellman var. tamatsuensis Miura (Bangiales, Rhodophyta). Aquaculture 274:126–131
Oliveira MC, Kurniawan J, Bird CJ et al (1994) A preliminary investigation of the order Bangiales (Bangiophycidae, Rhodophyta) based on the sequences of nuclear small-subunit ribosomal RNA genes. Phycol Res 43:71–79
Qiao LX, Liu HY, Guo BT et al (2007) Molecular identification of 16 Porphyra lines using sequence-related amplified polymorphism markers. Aquat Bot 87:203–208
Ragan MA, Bird CJ, Rice EL et al (1994) A molecular phylogeny of the marine red algae (Rhodophyta) based on the nuclear small-subunit rRNA gene. Proc Natl Acad Sci 91:7276–7280
Sahoo D, Tang XR, Yarish C (2002) Porphyra-the economic seaweed as a new experimental system. Curr Sci 83:1313–1316
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sun JW, Jin DM, Zhou CJ et al (2005) Identification of Porphyra lines (Rhodophyta) by AFLP DNA fingerprinting and molecular markers. Plant Mol Biol Rep 23:251–262
Sun JW, Jin DM, Weng ML et al (2006a) Development of SSR primers from EST sequences and their application in germplasm identification of Porphyra lines. Eur J Phycol 41:329–336
Sun X, Yang R, Liu BQ et al (2006b) Sequence analyses of rbcS and rbcL—rbcS intergenic spacer region in laver Porphyra haitanensis. Fish Sci 25(1):23–26
Tamura K, Dudley J, Nei M et al (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24(8):1596–1599
Thompson JD, Gibson TJ, Plewniak F et al (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25(24):4876–4882
Wang SJ, Zhang XP, Xu ZD et al (1986) Studies on the cultivation of the vegetative cells and protoplasts of Porphyra haitanensis Chang et Zheng. Ocean et Limnol Sinica 17(3):217–221
Woolcott GW, King RJ (1998) Porphyra and Bangia (Bangiaceae, Rhodophyta) in warm temperate waters of eastern Australia: morphological and molecular analyses. Phycol Res 46:111–123
Xie CT, Ji DH, Chen CS et al (2007) Sequence analysis and application of 5.8S rDNA and ITS regions of Porphyra haitanensis. Chin High Technol Lett 17(5):540–545
Xie CT, Chen CS, Ji DH et al (2008) Establishment of SRAP molecular marker reaction system for Porphyra haitanensis and genetic analyses of germplasm materials. Prog Nat Sci 18(3):247–253
Zhang XC, Qin S, Ma JH et al (eds) (2005) The genetics of marine algae. China agriculture press, Beijing
Acknowledgments
This research was supported in part by the National High Technology Research & Development Program of China (“863” Program) (Grant No. 2006AA10A413), the National Natural Science Foundation of China (Grant No. 40676077 and 40806065), the National Natural Science Foundation of Fujian Province, China (Grant No. 2007J0064), the Construction Program of Science and Technological Platform of Fujian Province, China (Grant No. 2007N2011), and the Foundation for Young Professors of Jimei University, China (Grant No. 2007C002).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, C., Xie, C., Ji, D. et al. Molecular divergence and application of the ITS-5.8S rDNA and RUBISCO spacer in Porphyra haitanensis Chang et Zheng (Bangiales, Rhodophyta). Aquacult Int 18, 1045–1060 (2010). https://doi.org/10.1007/s10499-010-9322-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10499-010-9322-y