
Abstract
Recent evidence have indicated that ferroptosis, a novel iron-dependent form of non-apoptotic cell death, plays a critical role in human cancers. Besides, emerging literatures have revealed the ovel function of N6-methyladenosine (m6A) in bladder cancer physiological. However, the underlying mechanism of m6A on bladder cancer is still unclear. Here, present work revealed that m6A methyltransferase (‘writer’) WTAP up-regulated in bladder cancer tissue and cells, indicating the poor prognosis of bladder cancer patients. Functionally, gain/loss-of-functional experiments illustrated that WTAP promoted the viability of bladder cancer cells and inhibited the erastin-induced ferroptosis. Mechanistically, there was a remarkable m6A modification site on 3’-UTR of endogenous antioxidant factor NRF2 RNA and WTAP could install its methylation. Moreover, m6A reader YTHDF1 recognized the m6A site on NRF2 mRNA and enhanced its mRNA stability. Therefore, these findings demonstrated potential therapeutic strategyies for bladder cancer via m6A-dependent manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bladder cancer acts as one of the most common tumors in the urinary system, ranking the 9th in global malignancy incidence and mortality [1, 2]. Clinically, surgery still functions as the mainstay therapeutic strategy for bladder cancer patients at localized stage [3, 4]. Particularly, the patients with local advanced or developed metastases will adopt surgical resection of the primary tumor [5]. After initial surgical treatments, increasing rate of bladder cancer patients relapse and targeted agents have been shown to reduce tumour recurrence and improve prognosis [6]. Therefore, the research on the bladder cancer pathological mechanism and new therapeutic target is still in the exploratory stage.
N6-methyladenosine (m6A) is a critical RNA modification type at post-transcriptional regulation, such as RNA stability, processing, localization, splicing and translation initiation [7,8,9]. There are three types of m6A key enzymes regulating m6A modification, including methyltransferase complex (‘writers’), demethylase (‘erasers’) and ‘reader’ [10]. Actually, m6A is installed by m6A methyltransferases complex, namely “writers”, including methyltransferase-like 3 (METTL3), METTL16, and WTAP. For example, METTL3 catalyzes the m6A modification in mRNAs [11]. Alternatively, the demethylases FTO and ALKBH5 remove the m6A from mRNAs to dynamically regulate their m6A modification [12]. Studies on m6A modification and biological function in mRNA have sprung up in recent years.
Ferroptosis is a novel type of iron-dependent programmed cell death, which is different from apoptosis, necrosis and autophagy [13,14,15]. Although ferroptosis is firstly reported in 2012 as a novel cell death form, the existing forms of cell death about iron and oxidative stress have been discovered for decades [16]. Ferroptosis could be repressed by the iron-chelating agent deferoxamine. Beyond the variational iron homeostasis, excessive generation of reactive nitrogen species or reactive oxygen species (ROS) also directly inspires the ferroptosis catalyzing the oxidation [17]. In human cancers, the pathological process of ferroptosis is frequently repressed by oncogenic factors. For example, in bladder cancer, fat mass and obesity-associated protein (FTO) expression significantly up-regulates in bladder cancer and is associated with its poor prognosis, and FTO overexpression promotes bladder cancer cells’ proliferation the FTO/miR-576/CDK6 pathways [18]. Thus, the potential roles of m6A in bladder cancer are increasing absorbing.
Previous studies have reported that m6A could regulate mRNA fate and play a critical role on bladder cancer development and progression. WTAP, as an important m6A writer, plays a critical role in human tumor, e.g. gastric cancer [19], colorectal cancer [20] and breast cancer [21]. Although some findings have shown that WTAP plays an essential role in the progression of tumors of urinary system, its deepgoing functions in bladder cancer are still unclear. Here, we found that m6A methyltransferase WTAP significantly up-regulated in the bladder cancer tissue and cells, and connected with the poor prognosis of bladder cancer patients. Moreover, WTAP accelerated the viability and repressed the erastin-induced ferroptosis (Fe2+ concentration, lipid ROS). Besides, WTAP installed the methylation on NRF2 mRNA, and m6A reader YTHDF1 recognized the m6A modification site on NRF2 mRNA and mediated its mRNA stability, forming a WTAP/m6A/YTHDF1/NRF2 axis.
Materials and methods
Clinical specimens
Clinically, tumor tissue and adjacent non-cancerous tissues (45 pairs samples) were both obtained from renal tumor patients who underwent nephrectomy at The First People’s Hospital of Xianyang from February 2019 to January 2021. All patients provided signed/informed consent. The study was approved by the Ethics Committees of The First People’s Hospital of Xianyang. For the immumohistochemical staining (IHC), Tissue sections were deparaffinized, rehydrated and heated in sodium citrate buffer for antigen retrieval. Then, slides were incubated with primary antibody, and examined under a fluorescence microscope (Carl Zeiss, Jena, Germany).
Cell lines and transfection
Human bladder epithelium cells (SV-HUC-1) and bladder cancer cell lines (J82, UM-UC-3) were obtained from cells were obtained from Shanghai Gene Chemistry (Shanghai, China). Cells were cultured with the Dulbecco’s Modified Eagle Medium (DMEM, cat. A1451801, Gibco, USA) containing 10% serum and 1% penicillin-streptomycin in an incubator containing 5% CO2 at 37 °C. Full length of WTAP plasmids and nontargeting sequence (negative control, NC) were synthesized (GenePharma, Shanghai, China). Cells were respectively transfected according to manufacture’s protocol. Other transfection was performed using lipofectamine 3,000 reagent (cat. L3000001, Invitrogen, Carlsbad, Calif, USA) when 80% confluence for cells. The transfection efficiency was detected by qRT-PCR.
RNA isolation and RT-qPCR
Total RNA was extracted from cultured cell samples or tissue samples by Trizol reagent (cat. A33254, Invitrogen, USA). To synthesize cDNA, RNA (1 µg) was reversely transcribed with the Primescript RT Reagent (TaKaRa, Japan) according to the manufacturer’s instructions. For quantitative analysis, qRT-PCR was conducted by SYBR® Premix Ex Taq™ Reagent (TaKaRa) on StepOne Plus Real-Time PCR system (Applied Biosystems, USA). The primers used for qRT-PCR were listed in Table S1. Relative mRNA expression was shown as fold changes and calculated by 2−ΔΔCt method normalized to β-actin.
Protein isolation and western blotting
Total cellular proteins were lysed by RIPA buffer (cat. R0278, Sigma, USA) containing protease inhibitors. The protein concentration was identified using BCA Protein Assay kit (Pierce, Thermo Scientific, cat. 23,225). Protein was separated using 10% SDS-PAGE gel and transferred to PVDF membrane (Millipore, USA). Blotting analysis was performed by standard procedure. After incubation on primary antibodies (anti-WTAP, anti-YTHDF1, Cell Signaling Technology, USA), PVDF membranes were incubated with peroxidase (HRP)-conjugated secondary antibodies. After washing twice, blots’ signals were detected using a chemiluminescence system (Bio-Rad, USA) and analyzed using Image Lab Software.
Colony formation and CCK-8 assay
For the colony formation assay, after 24 h transfection, bladder cancer cells in the logarithmic growth phase were trypsinized and then suspended in culture medium with 10% FBS. At density of 1000 cells/well, cells were re-seeded in six-well plates and cultured in a humidified atmosphere at 37 °C containing 5% CO2 in triplicate. After 2-weeks, colonies were washed with PBS and fixed with methanol. Then, colonies were stained with 0.1% crystal purple for 10 min and the mean colony numbers were calculated. For the CCK-8 assays, the transfected cells were treated by 0.25% trypsin, and then centrifuged. Cells were seeded into 96-well plate at 2500 cells/well in a 5% CO2 incubation at 37 C. After 24 h of incubation, cell counting kit-8 (CCK-8) reagent was added to the plates. The optical density (OD) value was detected by enzyme-mark reader (Multiskan FC, Waltham, MA, USA) at 450 nm. Each treatment group was assessed in triplicate.
Fe2+, GSH/GSSG analysis
The Fe2+ concentration was detected using the iron colorimetric assay kit (Applygen, Beijing, China, Cat. # E1042) referring to the manufacturer’s instructions under the ferrozine colorimetric method. The glutathione (GSH)/oxidized GSH (GSSG) ratio was detected using the GSH and GSSG assay kit (Beyotime Biotechnology, Jiangsu, China, Cat. # S0053) under the manufacturer’s instructions.
Cell death and lipid ROS measurement
The cell death was detected using flow cytometric analysis. For the lipid ROS level, bladder cancer cells were seeded in six-well plates and incubated with 5 µM C11-BODIPY (Thermo Fisher Scientific, Cat. #D3861) at 37 ◦C for 30 min. The cells were washed with PBS twice and digested with trypsin. Then, cells were suspended in serum-free medium (500 µL). The accumulated lipid ROS was measured by flow cytometric analysis on ID7000™ Spectral Cell Analyzer (Sony Biotechnology) and FlowJo software (Treestar).
RNA-binding protein immunoprecipitation (RIP)
To identify whether WTAP interacted with NRF2, RIP assays were performed using a Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Sigma Aldrich, cat. 17–704) according to the manufacturer’s protocol. In brief, the bladder cells were collected and lysed in radioimmunoprecipitation buffer (pH 7.5 20 mM Tris-HCl, 140 mM NaCl, 0.05% Triton X-100) containing protease inhibitor cocktail and RNase inhibitor. 5 µg of antibody was pre-bound to protein A/G magnetic beads in immunoprecipitation buffer for 2 h and incubated with cell lysate (100 µl) overnight. After incubation, RNA was eluted from beads by elution buffer (400 µl) for 2 h, precipitated was dissolved in RNase-free water and the enrichment was detected using real-time PCR.
Quantification of m6A in total RNA
Firstly, the total RNA was extracted from the indicated bladder cancer cells, and the m6A quantification was analyzed using the EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric, Epigentek, USA, Cat. # P-9005–48) following the manufacturer’s instructions.
Methylated RNA immunoprecipitation PCR (MeRIP-PCR)
To quantify the m6A-modified NRF2 level, MeRIP-PCR was performed using MeRIP m6A Kit (Merck Millipore, USA) according to the manufacture’s instruction. Total RNA was isolated from bladder cancer cells by Trizol. Anti-m6A antibody (3 µg, Millipore, Cat. # ABE572) and anti-IgG (3 µg, Cell Signaling Technology) were respectively conjugated to protein A/G magnetic beads in IP buffer overnight. IP buffer was supplemented with protease inhibitor and RNase inhibitor. For qRT-PCR assay, eluate or input total RNA was reverse-transcribed by Superscript III. The enrichment of m6A-containing transcripts was calculated by calculating the 2−ΔΔCt relative to the input sample.
Luciferase reporter assay
The wild-type or mutant sequences for m6A site of NRF2 3’-UTR was inserted in the vectors’ region. Both the pmirGLO-NRF2-3’UTR-WT and pmirGLO-NRF2-3’UTR-Mut were transfected into cells for 24 h, the firefly luciferase (F-luc) and Renilla luciferase (R-luc) were respectively detected by Dual Glo Luciferase Assay system (Promega, Madison, WI., USA). F-luc activity was normalized to R-luc activity. Promoter activity of NRF2 in cells was measured by luciferase assay according to protocols. After 24 h transfection and incubation, luciferase activity was detected using Dual Luciferase Reporter Assay kit (Promega, cat. E1910) according to the manufacturer’s instructions. R-luc normalized to F-luc activity was calculated.
RNA stability
J82 and UM-UC-3 cells were transfected with indicated plasmids, and treated with 5 µg/ml actinomyclin D (Act D, Merck, Darmstadt, Germany, cat. SBR00013) for 0, 3, 6 h. Then, total RNAs were isolated and harvested and then subjected to quantitative PCR analysis. The level of NRF2 mRNA was normalized to that of β-actin or GADPH control and the relative half-life of NRF2 mRNA was calculated.
Statistical analysis
Data were reported as mean ± standard deviation (SD) from three independent experiments’ data. For statistical analysis, two-tailed unpaired Student’s t-test was used for two groups’ difference and two-way ANOVA was used for multiple comparisons. Data analysis was carried out from SPSS 16.0 and GraphPad Prism software. P-value less than 0.05 was considered to statistical significance.
Results
WTAP indicated the poor prognosis for bladder cancer patients
Firstly, the expression of WTAP was detected in bladder cancer clinical samples. Results showed that WTAP levels increased in the tumour samples as comparing to normal tissues (Fig. 1 A). Moreover, the survival analysis showed that the patients with high-WTAP-level had a low survival rate, while the patients with low-WTAP-level had a high survival rate (Fig. 1B). In the cellular identification, results illustrated that the WTAP levels up-regulated in the bladder cancer cell lines (J82, UM-UC-3) corresponding to human bladder epithelium cells (SV-HUC-1) (Fig. 1 C). In tissue sample, the immumohistochemical staining (IHC) also indicated that WTAP up-regulated in the tumour samples as comparing to para-carcinoma tissue (Fig. 1D). Taken together, the data illustrated that WTAP was closely related to the poor prognosis for bladder cancer.

WTAP indicated the poor prognosis for bladder cancer. (A) Results by RT-PCR showed the WTAP levels in the clinical tumour samples as comparing to normal tissues samples. (B) The survival analysis showed the survival rate of bladder cancer patients with high-WTAP-level or low-WTAP-level. (C) RT-qPCR was performed to detect the WTAP level in human bladder epithelium cells (SV-HUC-1) and bladder cancer cell lines (J82, UM-UC-3). (D) Immumohistochemical staining (IHC) indicated the WTAP in the tumour samples as comparing to para-carcinoma tumor-adjacent tissue. The semi-quantitative analysis of WTAP was identified by Immunoreactivity score (IRS). *p < 0.05, **p < 0.01
WTAP promoted the proliferation of bladder cancer cells
Subsequently, to investigate the functions of WTAP on bladder cancer cells’ biological characteristic, gain/loss function assays were performed on bladder cancer cells (J82, UM-UC-3) using WTAP overexpression and silencing (Fig. 2 A). The transfection efficiency was detected using RT-PCR and western blot (Fig. 2B). For the viability of bladder cancer cells was detected using CCK-8 assay and clone formation assay. Clone formation assay showed that WTAP overexpression promoted the clone formation number and WTAP knockdown inhibited the clone formation number (Fig. 2 C). CCK-8 assay revealed that WTAP overexpression promoted the proliferative ability and WTAP knockdown inhibited the proliferative ability (Fig. 2D). Taken together, the data illustrated that WTAP promoted the proliferation of bladder cancer cells.

WTAP promoted the proliferation of bladder cancer cells. (A) Gain/loss function assays were performed using WTAP overexpression (in J82 cells) and silencing (in UM-UC-3 cells). The WTAP mRNA level was detected using RT-PCR. (B) The transfection efficiency was detected using RT-PCR and western blot in bladder cancer cells (J82, UM-UC-3). (C) Clone formation assay was performed to display the clone formation number with WTAP overexpression (in J82 cells) or WTAP knockdown (in UM-UC-3 cells). (D) CCK-8 assay was performed to display the proliferative ability with WTAP overexpression (in J82 cells) or WTAP knockdown (in UM-UC-3 cells). *p < 0.05, **p < 0.01
WTAP reduced erastin-induced ferroptosis in bladder cancer
Ferroptosis is characterized by the accumulation of iron-dependent lipid peroxides (lipid-ROS), which leads to lethal cellular damage. Ferroptosis process could be divided into ROS production (upstream) and ferroptosis execution (downstream). Here, in this research, ferroptosis was induced using erastin, and data showed that cell death was increased upon erastin treatment (Fig. 3 A, 3B). Meanwhile, WTAP overexpression repressed the cell death and WTAP silencing increased the cell death. Given that Fe2+, GSH/GSSG, and lipid ROS were essential for ferroptosis process, we further measured the concentrations in erastin-treated bladder cancer cells. Fe2+ concentration analysis revealed that WTAP overexpression inhibited the Fe2+ level, while WTAP silencing up-regulated the Fe2+ level (Fig. 3 C). Then, GSH/GSSG analysis revealed that WTAP overexpression up-regulated the GSH/GSSG level, and WTAP silencing decreased the GSH/GSSG level (Fig. 3D). Lipid ROS analysis found that WTAP overexpression inhibited the lipid ROS level, while WTAP silencing increased the lipid ROS level (Fig. 3E F). Taken together, the data illustrated that WTAP reduced erastin-induced ferroptosis in bladder cancer.

WTAP reduced erastin-induced ferroptosis in bladder cancer. (A, B) The cell death was detected using flow cytometry in bladder cancer with erastin treatment using WTAP overexpression (in J82 cells) and silencing (in UM-UC-3 cells). (C) Fe2+ concentration analysis was performed with WTAP overexpression (in J82 cells) and silencing (in UM-UC-3 cells). (D) GSH/GSSG analysis was performed in bladder cancer cells with WTAP overexpression (J82 cells) and silencing (UM-UC-3 cells). (E, F) Flow cytometry was performed to detect the lipid ROS level in bladder cancer cells with WTAP overexpression (in J82 cells) and silencing (in UM-UC-3 cells). *p < 0.05, **p < 0.01
NRF2 acted as the target of WTAP via m6A-dependent manner
Given our work found that WTAP could regulate the ferroptosis of bladder cancer cells, we further screened the downstream molecular to discovery the target of WTAP. NRF2 (also known as NFE2L2) was a critical element for tumour ferroptosis, and we found that there was a remarkable m6A modification site on NRF2 mRNA 3’-UTR (Fig. 4 A). The m6A modification concentration analysis showed that the m6A modification level increased in bladder cancer cells, suggesting the high-m6A enrichment in bladder cancer (Fig. 4B). Through the RMBase v2.0 (https://rna.sysu.edu.cn/rmbase/), we found the m6A modification motif is AGGAC (Fig. 4 C). Moreover, the more precise location of m6A site on the 3’-UTR of NRF2 mRNA (Fig. 4D). In bladder cancer cells, the NRF2 mRNA level also up-regulated (Fig. 4E). Besides, NRF2 mRNA level was increased in WTAP overexpression transfection, which was reduced in WTAP silencing (Fig S1). The RIP-PCR analysis showed that NRF2 mRNA was significantly enriched by anti-m6A antibody as comparing to controls (Fig. 4 F). Moreover, the RIP-PCR analysis demonstrated that WTAP overexpression up-regulated the enrichment of NRF2 mRNA by anti-m6A antibody, and WTAP silencing reduced the enrichment (Fig. 4G). Taken together, the data illustrated that NRF2 acted as the target of WTAP via m6A-dependent manner.

NRF2 acted as the target of WTAP via m6A-dependent manner. (A) SRAMP (http://www.cuilab.cn/sramp) showed that there was a remarkable m6A modification site in NRF2 mRNA 3’-UTR. (B) The m6A modification concentration analysis was detected in bladder cancer cells. (C) RMBase v2.0 (https://rna.sysu.edu.cn/rmbase/) showed the m6A modification motif is AGGAC. (D) The more precise location of m6A site on the 3’-UTR of NRF2 mRNA. (E) Western blot and RT-PCR were conducted to measure the NRF2 protein and mRNA level in bladder cancer cells. (F) The RIP-PCR analysis was performed to show the NRF2 mRNA enrichment by anti-m6A antibody or controls. (G) The RIP-PCR analysis was performed to demonstrate NRF2 mRNA level by anti-m6A antibody or anti-IgG antibody. *p < 0.05, **p < 0.01
WTAP enhanced the stability of NRF2 mRNA via an YTHDF1-m6A-dependent pattern
Given that NRF2 mRNA acted as the downstream target of WTAP, we subsequently detected the possible pathway mediated this regulation. In the classical research, the function of m6A methyltransferase is always mediated by m6A reader, thus, we screened the YTHDFs to identify which reader mediated it. Results showed that YTHDF1 showed remarkable up-regulation in tumour cells, thereby acting as the potential m6A reader for WTAP-NRF2 mRNA (Fig. 5A). The RIP-PCR analysis showed that NRF2 mRNA was significantly enriched by anti-YTHDF1 antibody as comparing to controls (Fig. 5B). Moreover, the RIP-PCR analysis demonstrated that WTAP overexpression up-regulated the enrichment of NRF2 mRNA by anti-YTHDF1 antibody, and WTAP silencing reduced the enrichment (Fig. 5 C, 5D). The correlation analysis with in NRF2 (NFE2L2) and WTAP (Fig. 5E), or NRF2 (NFE2L2) and YTHDF1 (Fig. 5F) was identified, showing a positive correlation both within them. Then, luciferase reporter assay using wild-type (WT) or mutant (Mut) of NRF2 mRNA 3’-UTR sequence indicated that m6A maintained the mRNA stability of NRF2 by WTAP in bladder cancer (Fig. 5G H). RNA decay analysis revealed that WTAP overexpression enhanced NRF2 remaining level upon Act D treatment, while WTAP silencing reduced the NRF2 remaining level (Fig. 5I). Moreover, RNA decay analysis also demonstrated that YTHDF1 knockdown repressed the NRF2 mRNA level, while WTAP overexpression reversed the NRF2 mRNA level in co-transfection of si-YTHDF1 and oe-WTAP (Fig. 5 J). Results indicated that WTAP/TYHDF1 enhanced NRF2 mRNA stability mainly through the m6A-dependent manner. Besides, NRF2 mRNA level was reduced with YTHDF1 silencing (Fig S2). Taken together, the data illustrated that WTAP enhanced the stability of NRF2 mRNA via an YTHDF1-m6A-dependent pattern.

WTAP enhanced the stability of NRF2 mRNA via an YTHDF1-m6A-dependent pattern. (A) RT-PCR was performed to detect YTHDFs’ mRNA levels in bladder cancer cells. (B) The RIP-PCR analysis using anti-YTHDF1 antibody or control anti-IgG antibody displayed the enrichment of NRF2 mRNA. (C, D) RIP-PCR analysis was performed using anti-YTHDF1 in bladder cancer cells with WTAP overexpression (in J82 cells) and silencing (in UM-UC-3 cells). The NRF2 mRNA level was detected using RT-PCR. (E) The correlation analysis within NRF2 (NFE2L2) and WTAP was identified by GEPIA (http://gepia.cancer-pku.cn/index.html). (F) The correlation analysis within NRF2 (NFE2L2) and YTHDF1 was identified by GEPIA (http://gepia.cancer-pku.cn/index.html). (G) The schematic diagram for wild-type (WT) or mutant (Mut) of NRF2 mRNA 3’-UTR sequence for luciferase reporter assay. (H) Luciferase reporter assay was performed to show the luciferase activity. Relative luciferase activity was computed by the ratio of Firefly and Renilla luciferase values. (I) RNA decay analysis was conducted to reflect the NRF2 remaining level upon Act D treatment in bladder cancer with WTAP overexpression (in J82 cells) and silencing (in UM-UC-3 cells). (J) RNA decay analysis also demonstrated the NRF2 mRNA level in co-transfection of si-YTHDF1 and oe-WTAP. *p < 0.05, **p < 0.01

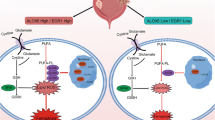
m 6 A writer WTAP targets NRF2 to accelerate bladder cancer malignancy via YTHDF1/NRF2/m6A-dependent ferroptosis regulation
Fig S1. RT-PCR was performed to detect NRF2 mRNA level in J82 cells with WTAP overexpression transfection, and in UM-UC-3 cells with WTAP silencing
Fig S2. RT-PCR was performed to detect NRF2 mRNA level in J82 cells with si-YTHDF1.
Discussion
Ferroptosis is a special mode of death found in a high-throughput small-molecule screening, which could be blocked by antioxidants or iron chelating agents (desferriamine-mesylate). Ferroptosis is characterized by lipid peroxidation, iron and reactive oxygen species (ROS) dependent manner. Ferroptosis is known to be involved in a variety of biological metabolism and pathological processes in vivo, and is also widely involved in the formation and development of malignant tumors, playing a key regulatory role in the tumour microenvironment. In bladder cancer, increasing evidence had indicated that ferroptosis participates in the tumor progression [22].
Here, we found that m6A writer WTAP up-regulated in the bladder cancer tumorigenesis and functioned as an oncogene in this pathophysiological process. The high-expression of WTAP in bladder cancer tissue/cells and the poor prognosis both indicated the risk factor for WTAP in bladder cancer. Functionally, WTAP promoted the proliferation of bladder cancer cells. Moreover, results revealed that WTAP repressed the erastin-induced ferroptosis (increasing cell death rate, Fe2+, lipid ROS, and reducing GSH/GSSG). The findings of WTAP on bladder cancer illustrated that WTAP could target the ferroptosis-related physiopathologic phenotype.
The ferroptosis on bladder cancer is a more and more valuable research hotspot. Through regulating ferroptosis, various of elements could modulate the bladder cancer progression. For example, ferroptosis inducer Fin56 triggers the ferroptosis by promoting glutathione peroxidase 4 (GPX4) protein degradation depends on the autophagic machinery, supporting the concept that ferroptosis is a type of autophagy-dependent cell death [23]. Besides, tumor mutation burden is positively correlated to the ferroptosis score and the low ferroptosis score is related to high-response to immunotherapy by PD-1 blockage, indicating a positive correlation between cisplatin chemotherapy sensitivity and ferroptosis score [24].
Regarding to the deepgoing mechanism for WTAP regulating ladder cancer’s ferroptosis., we aimed to investigate the molecular mechanism. Results indicated that there was a remarkable m6A modification site in NRF2 (also known as NFE2L2) mRNA 3’-UTR. NRF2 is a critical element for tumour ferroptosis, and we found that NRF2 acted as the target of WTAP via m6A-dependent manner. The m6A modification concentration showed that the m6A modification level increased in bladder cancer cells, suggesting the high-m6A modification in bladder cancer. RIP-PCR analysis revealed that WTAP overexpression up-regulated the enrichment of NRF2 mRNA by anti-m6A antibody. Therefore, we concluded that WTAP significantly combined with NRF2 through molecular interaction.
Increasing evidence has showed that m6A modification could regulate the fate of RNA, including nuclear translocation, splicing, stability and degradation. Here, we tried to investigate how WTAP regulated the NRF2 mRNA feature. Interestingly, we found that WTAP enhanced the NRF2 mRNA stability via targeting m6A modification site. We were not just limited to this discovery, and then identified whether other m6A elements involved in this course. In classical research, the functions of m6A methyltransferase are always mediated by m6A readers, thus, we screened the YTHDFs to identify which reader mediated it. Results showed that YTHDF1 exerted more remarkable up-regulation in tumour cells, thereby acting as the potential m6A reader for WTAP-NRF2 mRNA.
In the bladder cancer, the potential role of NRF2 (Nuclear factor (erythroid-derived 2)-like 2) is increasingly valued. In bladder cancer patients’ peripheral blood leukocytes, the expression of NRF2 and NRF2-modulated genes up-regulated [25]. It has identified that the p62-KEAP1-NRF2 pathway promotes bladder cancer tumor growth and tumorigenesis via NRF2-dependent antioxidative response [26]. Thus, the critical function of NRF2 in bladder cancer has important research value.
Taken together, the present research revealed novel findings about WTAP in bladder cancer. The high-expression of bladder cancer was also connected with the poor prognosis of bladder cancer patients, and accelerated the proliferation and repressed the erastin-induced ferroptosis. Mechanistically, WTAP installed the methylation on NRF2 mRNA and m6A reader YTHDF1 recognized the m6A modification to mediate its mRNA stability, forming a WTAP/m6A/YTHDF1/NRF2 axis. Based on these findings, more precise targeted therapy could be developed.
Data Availability
Not Applicable.
References
Dobruch J, Daneshmand S, Fisch M, Lotan Y, Noon AP, Resnick MJ et al (2016) Gender and bladder Cancer: a collaborative review of etiology, Biology, and outcomes. Eur Urol 69:300–310
Dobruch J, Oszczudłowski M (2021) ; 57
Lenis AT, Lec PM, Chamie K, Mshs MD (2020) Bladder Cancer: A Review Jama 324:1980–1991
Martinez Rodriguez RH, Buisan Rueda O, Ibarz L (2017) Bladder cancer: Present and future. Med Clin 149:449–455
Ng K, Stenzl A, Sharma A, Vasdev N (2021) Urinary biomarkers in bladder cancer: a review of the current landscape and future directions. Urol Oncol 39:41–51
Siracusano S, Rizzetto R, Porcaro AB (2020) Bladder cancer genomics Urologia 87:49–56
An Y, Duan H (2022) The role of m6A RNA methylation in cancer metabolism. Mol Cancer 21:14
Cai X, Liang C, Zhang M, Xu Y, Weng Y, Li X et al (2022) N6-methyladenosine modification and metabolic reprogramming of digestive system malignancies. Cancer Lett 544:215815
Chang H, Yang J, Wang Q, Zhao J, Zhu R (2022) Role of N6-methyladenosine modification in pathogenesis of ischemic stroke. Expert Rev Mol Diagn 22:295–303
Deng LJ, Deng WQ, Fan SR, Chen MF, Qi M, Lyu WY et al (2022) m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol Cancer 21:52
Liu L, Li H, Hu D, Wang Y, Shao W, Zhong J et al (2022) Insights into N6-methyladenosine and programmed cell death in cancer. Mol Cancer 21:32
Zhang N, Ding C, Zuo Y, Peng Y, Zuo L (2022) N6-methyladenosine and neurological Diseases. Mol Neurobiol 59:1925–1937
Chen X, Li J, Kang R, Klionsky DJ, Tang D (2021) Ferroptosis: machinery and regulation. Autophagy 17:2054–2081
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N et al (2020) Ferroptosis: past, present and future. Cell Death Dis 11:88
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C et al (2019) Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol 12:34
Tang D, Chen X, Kang R, Kroemer G (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31:107–125
Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J et al (2020) Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol 13:110
Zhou G, Yan K, Liu J, Gao L, Jiang X, Fan Y (2021) FTO promotes tumour proliferation in bladder cancer via the FTO/miR-576/CDK6 axis in an m6A-dependent manner. Cell death discovery 7:329
Yang D, Chang S, Li F, Ma M, Yang J, Lv X et al (2021) M(6) a transferase KIAA1429-stabilized LINC00958 accelerates gastric cancer aerobic glycolysis through targeting GLUT1. IUBMB Life 73:1325–1333
Zhou Y, Pei Z, Maimaiti A, Zheng L, Zhu Z, Tian M et al (2022) M(6)a methyltransferase KIAA1429 acts as an oncogenic factor in colorectal cancer by regulating SIRT1 in an m(6)A-dependent manner. Cell death discovery 8:83
Zhang X, Dai XY, Qian JY, Xu F, Wang ZW, Xia T et al (2022) SMC1A regulated by KIAA1429 in m6A-independent manner promotes EMT progress in breast cancer. Mol therapy Nucleic acids 27:133–146
Yi K, Liu J, Rong Y, Wang C, Tang X, Zhang X et al (2021) Biological Functions and Prognostic Value of Ferroptosis-Related genes in bladder Cancer. Front Mol Biosci 8:631152
Sun Y, Berleth N, Wu W, Schlütermann D, Deitersen J, Stuhldreier F et al (2021) Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells. Cell Death Dis 12:1028
Xia QD, Sun JX, Liu CQ, Xu JZ, An Y, Xu MY et al (2022) Ferroptosis patterns and Tumor Microenvironment Infiltration characterization in bladder Cancer. Front cell Dev biology 10:832892
Reszka E, Jablonowski Z, Wieczorek E, Gromadzinska J, Jablonska E, Sosnowski M et al (2013) Expression of NRF2 and NRF2-modulated genes in peripheral blood leukocytes of bladder cancer males. Neoplasma 60:123–128
Li T, Jiang D, Wu K (2020) p62 promotes bladder cancer cell growth by activating KEAP1/NRF2-dependent antioxidative response. Cancer Sci 111:1156–1164
Acknowledgements
No.
Funding
Declaration.
Not Applicable.
Author information
Authors and Affiliations
Contributions
Ke Wang, Gang Wang, Gang Li, Wei Zhang, Yarong Wang wrote the main manuscript text. Xiaofeng Lin, Chengxian Han, Hanxuan Chen, Liang Shi, Abudoula Reheman, Jingkai Li, Zhaomin, Xinxuan Yang prepared Figs. 1, 2, 3, 4, 5 and 6. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.


Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
Cite this article
Wang, K., Wang, G., Li, G. et al. m6A writer WTAP targets NRF2 to accelerate bladder cancer malignancy via m6A-dependent ferroptosis regulation. Apoptosis 28, 627–638 (2023). https://doi.org/10.1007/s10495-023-01817-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-023-01817-5