
Abstract
Ferroptosis is an iron-dependent and phospholipid peroxidation-mediated cell death, which has been identified to be involved in sepsis-induced injury. However, the in-depth molecular mechanisms of N6-methyladenosine (m6A) and ferroptosis on sepsis-induced myocardial injury are still unclear. Here, in the septic myocardial injury, m6A methyltransferase METTL3 level and methylation level high-expressed in lipopolysaccharide (LPS)-induced cardiomyocytes (H9C2). Functionally, METTL3 silencing repressed the ferroptosis phenotype induced by LPS. Mechanistically, METTL3-mediated m6A methylation on solute carrier family 7 member 11 (SLC7A11) empowered its mRNA with high methylation level. Moreover, YTHDF2 directly bound to the m6A modification sites of SLC7A11 to mediate the mRNA degradation. The m6A modified SLC7A11 mRNA was recognized by YTHDF2, which promoted the decay of SLC7A11 mRNA, consequently up-regulating ferroptosis in sepsis-induced myocardial injury. Together, these findings establish a role of METTL3 in the ferroptosis of LPS-induced cardiomyocytes, and provide potential therapeutic target to treat the sepsis-induced myocardial injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In clinic treatment, sepsis is a leading cause of morbidity/mortality after surgery and could give rise to multiple organ failure [1, 2]. Sepsis-induced myocardial injury is a common clinical complication because heart is one of the most vulnerable target organs in sepsis pathophysiological process [3]. The clinical manifestations of sepsis-induced myocardial injury include degeneration and necrosis of cardiomyocytes, myocardial contraction and diastolic dysfunction [4, 5]. In addition to timely clinical treatment, it is also essential to investigate the molecular mechanism for sepsis-induced myocardial injury.
N6-methyladenosine (m6A), as one of the most remarkable RNA post-transcription modification pattern, has been identified as critical determining factor in human pathological physiology [6, 7]. In present progress, there are multiple elements having been verified in sepsis, for example m6A demethylase ALKBH5 [8], m6A methylase METTL3 [9] and YTHDF1 [10]. In the regulation of m6A modification, regulators (methyltransferase ‘writer’, demethylase ‘eraser’, and binding protein ‘reader’) exert their functions by directly or indirectly modification [11, 12].
Ferroptosis is new type of programmed cell death and an iron-dependent programmed death, which has been identified to participate in numerous human tumor progressions. Ferroptosis is different from apoptosis, necrosis, pyroptosis and autophagy, which is a new mode of death induced by iron-dependent lipid peroxidation. Ferroptosis has been reported to regulate series of diseases and physiopathology. For example, in atherosclerosis cardiovascular diseases or sepsis-induced cardiomyopathy, ferroptosis plays an important regulatory role in the occurrence and development of many heart diseases, as well as myocardial ischemia/reperfusion injury, cardiomyopathy and heart failure [13]. Therefore, the role of ferroptosis in cardiovascular diseases has a very important function.
In the pathophysiology of ferroptosis, SLC7A11 plays a critical function. SLC7A11 (also commonly known as xCT) is a cystine/glutamate antiporter and import cystine for glutathione biosynthesis and antioxidant defense. In the sepsis-induced cardiac injury, LPS decreases the level of Gpx4 and SLC7A11 and increases the expression of P53 and ferritin [14]. Furthermore, ferrostatin-1, a specific ferroptosis inhibitor, protect against LPS-induced cardiac injury. Thus, these findings illustrate the critical function on LPS-induced cardiac injury.
Here, we reported a novel insight of METTL3 on ferroptosis in sepsis-induced myocardial injury, providing a regulatory by m6A-dependent manner. In the sepsis inspired pathophysiological process, METTL3 level increased upon LPS treatment, as well as the ferroptosis grade. Through YTHDF2-dependent mRNA decay progression, METTL3 installed the methylation level of SLC7A11 mRNA and then YTHDF2 accelerated the degradation, thereby reducing SLC7A11 protein level and induced ferroptosis in LPS-induced H9C2 cells.
Materials and methods
Cells culture and treatment
Rat cardiomyocyte (H9C2) cells were purchased with ScienCell (Catalog #R6200, ScienCell, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% penicillin-streptomycin (Solarbio) and 10–12% FBS (fetal bovine serum, Gibco, Gran Island, NY, USA) with 5% CO2 in 37 °C incubator. When cells were developed to 80% confluence, medium was refreshed. Meanwhile, for sepsis cellular model construction, H9C2 cells were administered with increasing LPS doses (0, 1, 2, 5, 10 µg/mL) for 24 h.
Transfection of plasmids
For stable METTL3 silencing transfection, shRNAs targeting METTL3 were synthesized by Ribobio (Ribobio, Guangzhou, China) and inserted into PLKO.1 plasmids. To overexpress YTHDF2, its full length of YTHDF2 genome were amplified from the cDNA and cloned into vector pmiRGLO (Promega, Madison, WI, USA). The negative vector acted as the control groups and also be generated. All constructed sequences were synthesized by RiboBio (Guangzhou, China) and confirmed by sequencing. Cardiomyocytes were planted in six-well plates 24 h, and after 24 h of LPS (10 µg/mL) treatment, cardiomyocytes (H9C2) were transfected following protocols’ instruction with 50–60% confluence using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacture instructions. The sequences that were used were shown in Table S1.
RNA isolation and qRT-PCR
For RNA qRT-PCR analysis, RNA isolation was extracted from cells using MiniBEST Universal RNA Extraction Kit (TaKaRa, Japan). Reverse transcription was performed by one-step PrimeScript RTPCR kit using 1 µg of total RNA. Quantitative PCR was performed by TB Green® Fast qPCR Mix (TaKaRa, Japan) with three repeated reactions. Using the ddCt method to compare with the GAPDH/actin level, the relative mRNA expression level was calculated/normalized to the control sample. Primers were listed in Supplementary Table S1.
Western blot
Total protein samples from myocardial cells were prepared by radioimmunoprecipitation assay (RIPA) lysis buffer (cat. BYL40825, JRDUN Biotechnology Co. Ltd, Shanghai, China;). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to isolate protein samples, and the protein was electro-transferred to nitrocellulose membranes (PVDF) followed by blocking with 5% skim milk at room temperature for 2 h. The membranes were incubated with primary antibodies against METTL3 (Abcam, 1:1000, # ab195352), against YTHDF2 (Cell Signaling Technology, 1:1000, #71,283) and beta-actin (Cell Signaling Technology, #4970) at 4 °C overnight. The western blotting bands were determined using ImageJ software.
Cell counting Kit-8 viability analysis
After transfection, cells at logarithmic growth phase were washed twice with PBS. Cells were seeded into a 96-well plate (2 × 103 cells in each well) in 5% CO2 at 37 °C. After 24 h, cell counting kit-8 (CCK-8) kit (Dojindo Laboratories, Kumamoto, Japan) was added to cells (10 mL/well). After the reaction, the optical density (OD) value of wells was detected at 450 nm using an automatic enzyme-mark reader.
Lipid ROS and cell death analysis
The lipid ROS and cell death for H9C2 cells were calculated using flow cytometry according to published protocols [15]. In brief, H9C2 cells were stained with BODIPY™ 581/591 C11 (Cat. D3861, Invitrogen Thermo-Fisher) for lipid ROS analysis. Besides, the cells were stained with Invitrogen™ propidium iodide (cat. P1304MP, 1:1000; Invitrogen) for 30 min at 37 °C for cell death rate.
GSH/oxidized GSH (GSSG) ratio, Fe2+, MDA analysis
The cellular glutathione (GSH)/oxidized GSH (GSSG) ratio was detected using GSH and GSSG assay kit (cat. S0053, Beyotime Biotechnology, Jiangsu, China) according to the manufacturer’s instructions [16]. The malondialdehyde (MDA) quantitative detection was identified using Lipid Peroxidation MDA Assay Kit (cat. S0131S, Beyotime Biotechnology, Jiangsu, China). The iron load assay was calculated using Iron Colorimetric Assay kit (Cat. ab83366, Abcam, USA) at absorbance 550 nm with microplate reader.
RNA immune‑precipitation (RIP)
The RIP assay was performed using Magna RIP™ RNA Binding Protein Immunoprecipitation Kit (cat. 17–704, Millipore, USA) according to the manufacturer’s instructions using protein A/G magnetic beads. In brief, cell lysate was collected/incubated with magnetic bead protein A/G coated with IgG antibody (5 µg, Beyotime, cat. A0208), anti-m6A antibody (Synaptic Systems, cat. 202,003), anti-YTHDF2 antibody (Cat No. 22803-1-AP) at 4 °C overnight. Incubation with goat anti-rabbit IgG (Beyotime, Shanghai, China, cat. A0208) was subsequently performed for 1 h at room temperature. The RNA was purified and extracted using the RIPA. The relative expression of SLC7A11 was detected by RT-qPCR.
MeRIP-qPCR
Total RNA (2 mg) and m6A mixture were added to 300 ml of IP buffer (pH 7.4, 50 mM Tris-HCl, 0.1% NP40, 150 mM NaCl, and 40 U/ml RNase inhibitor) and then incubated with anti-m6A rabbit polyclonal antibody (2 mg, Synaptic Systems) as previously described [17]. Anti-rabbit IgG suspension was blocked with bovine serum albumin (0.5%) at 4 °C for 2 h. Being washed with IP buffer, the resuspended in the total RNA-antibody mixture. After elution, beads were washed at 50 °C for 1 h and followed by centrifugation at 4 °C of 14,000 rpm for 10 min. After precipitated RNA extracted, relative RNA level was detected by qRT-PCR.
m6A content analysis
Total RNA was extracted sing TRIzol reagent (Invitrogen, USA). Poly(A) RNA was purified by GenElute mRNA Miniprep Kit (Sigma, cat. # MRN10) [18]. m6A content was assayed by the m6A RNA Methylation Assay Kit (Abcam; # ab185912). In brief, binding solution (80 µL) and sample RNA (200 ng) were added into each well, and then incubated for 90 min at 37 °C. After RNA binding, each well was washed three times with washing buffer. At room temperature of 60 min, each well was incubated with anti-m6A antibody (Abcam; #ab208577) at room temperature for 30 min in the dark for 1–10 min at 25 °C. Reaction was determined using a microplate reader at 450 nm wavelength.
Luciferase reporter assays
The dual-luciferase reporter gene vector for SLC7A11 target gene with wild type or mutants were formulated respectively named as Luc-SLC7A11-WT and Luc-SLC7A11-Mut as previously described [16]. The two reporter plasmids, sh-METTL3 and sh-NC plasmids, were co-transfected into 293T cells, and then lysed for 36 h. After transfection and centrifuged of 1 min/12,000 rpm, the supernatant was collected, and the luciferase activity was detected using Dual-Luciferase® Reporter Assay System (Promega). Then, firefly luciferase solution was added into samples to detect the firefly luminescence Renilla luciferase.
RNA decay or mRNA stability measurement
H9C2 cells were treated by actinomycin D (Act D, 2 µg/ml, GlpBio, Montclair, CA, USA; #GC16866) for 30 min and acted as 0 h samples [19]. The 3 and 6 h samples were then collected for the total mRNA extraction. cDNA synthesis by reversely transcribed and then subsequently detected using an oligo(dT) primer. The quantitated RNA level was determined by RT-qPCR.
Statistical analysis
All data in present study were processed by GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA) and expressed as mean ± standard deviation (SD). For in vitro experiments, Student’s t-tests were used to calculate the p-value. P-value < 0.05 was considered statistically significant.
Results
The m6A modification profile and ferroptosis condition in LPS-treated cardiomyocytes
In the previous studies [9], we performed the Methylated RNA immunoprecipitation sequencing (MeRIP-Seq) to discovery the m6A profile in LPS-treated cardiomyocytes. In the LPS-induced H9C2 cells, candidate motifs were screened, e.g. AAGGACU (Fig. 1A). MeRIP-Seq revealed the m6A modification distribution in myocardial cell injury cardiomyocytes, including 3′-UTR, 5′-UTR and CDS (Fig. 1B). To construct the model of septic myocardial cell injury, the cardiomyocytes (H9C2) was induced by LPS (10 µg/mL). In the treated cardiomyocytes, the m6A modification level increased with concentration dependent manner (0, 1, 2, 5, 10 µg/mL) (Fig. 1C). Moreover, the METTL3 protein expression also increased with the increasing doses of LPS (Fig. 1D). The lipid ROS expression was calculated by flow cytometry and resulted illustrated that the ROS level significantly up-regulated with LPS concentration (Fig. 1E). Moreover, the GSH level was remarkably decreased in cardiomyocytes (Fig. 1F). Taken together, these findings showed the m6A modification profile and ferroptosis condition in LPS-treated cardiomyocytes.

The m6A modification profile and ferroptosis condition in LPS-treated cardiomyocytes. A Methylated RNA immunoprecipitation sequencing (MeRIP-Seq) was performed to discovery the m6A profile in LPS-treated cardiomyocytes. The candidate motifs were screened, e.g. AAGGACU. B The m6A modification distribution in myocardial cell injury cardiomyocytes, including 3’-UTR, 5’-UTR and CDS. C The m6A modification analysis showed the m6A modification level with concentration dependent manner (0, 1, 2, 5, 10 µg/mL) in the LPS induced cardiomyocytes (H9C2). D Western blot shoed the METTL3 protein expression with the increasing doses of LPS. E Flow cytometry was performed to detect the lipid ROS expression. F The GSH level was calculated in cardiomyocytes. Cellular experiments were performed in triplicate and were repeated three times. **p < 0.01; *p < 0.05
METTL3 knockdown alleviated LPS-induced ferroptosis in cardiomyocytes
The cellular phenotype of cardiomyocytes was determined in LPS (10 µg/mL) induced cardiomyocytes (H9C2). Firstly, the knockdown of METTL3 was constructed using the short hairpin RNA (shRNA-METTL3, sh-METTL3), and the transfected efficient was detected using RT-PCR (Fig. 2A) and western blot (Fig. 2B). For the viability, the knockdown of METTL3 recovered the proliferative ability under LPS treatment (Fig. 2C). For the cell death, the knockdown of METTL3 reduced the death rate induced by LPS treatment in H9C2 cells (Fig. 2D). For the lipid ROS, the knockdown of METTL3 repressed the lipid ROS of cardiomyocytes (H9C2) under LPS treatment (Fig. 2E, F). The GSH/GSSG analysis revealed that knockdown of METTL3 up-regulated the GSH/GSSG rate (Fig. 2G). The iron load assay revealed that knockdown of METTL3 down-regulated the Fe2+ concentration upon LPS treatment (Fig. 2 H). The malondialdehyde (MDA) quantitative analysis revealed that knockdown of METTL3 inhibited the MDA level (Fig. 2I). Overall, these data indicated that METTL3 knockdown alleviated LPS-induced ferroptosis in cardiomyocytes.

METTL3 knockdown alleviated LPS-induced ferroptosis in cardiomyocytes. A RT-PCR and B western blot were performed to detect the transfected efficient of METTL3 knockdown using the short hairpin RNA (shRNA-METTL3, sh-METTL3) in LPS-induced ferroptosis in cardiomyocytes (H9C2). C CCK-8 assay was performed to detect the proliferative ability of cardiomyocytes under LPS treatment with sh-METTL3 or sh-NC. D The cell death was performed to detect the death rate of LPS administered H9C2 cells. E, F Flow cytometry was performed to detect the lipid ROS of cardiomyocytes (H9C2) under LPS treatment. G The GSH/GSSG analysis was performed to detect the GSH/GSSG rate of cardiomyocytes (H9C2) under LPS treatment. H The iron load assay was performed to detect the Fe2+ concentration under LPS treatment. I The malondialdehyde (MDA) quantitative analysis was performed to detect the MDA level upon knockdown of METTL3. Cellular experiments were performed in triplicate and were repeated three times. **p < 0.01; *p < 0.05
SLC7A11 functioned as a target of METTL3
Using the m6A seq results, we found that there was potential m6A modification in the SLC7A11 mRNA (Fig. 3A). In the 3′-UTR of SLC7A11 mRNA, the suitable m6A modified site was ‘GGACT’ (Fig. 3B). Using the online tool (SRAMP, http://www.cuilab.cn/sramp), we found that the m6A modified site in the SLC7A11 genome (Fig. 3C). In the cardiomyocytes (H9C2) under LPS treatment, the protein level of SLC7A11 decreased (Fig. 3D). For the molecular binding within SLC7A11 and METTL3, the RIP-PCR analysis was performed and results indicated that METTL3 significantly correlated to SLC7A11 (Fig. 3E). For the m6A modification level of SLC7A11, MeRIP-PCR was performed and results indicated that METTL3 knockdown repressed the m6A modification level of SLC7A11 mRNA in cardiomyocytes under LPS treatment (Fig. 3F). Overall, these data indicated that SLC7A11 functioned as a target of METTL3.

SLC7A11 functioned as a target of METTL3. A The potential m6A modification site in the SLC7A11 mRNA and corresponding m6A motif. B The suitable m6A modified site in the SLC7A11 mRNA was ‘GGACT’ in its 3′-UTR. C The m6A modified site in the SLC7A11 genome was shown by the online tool (SRAMP, http://www.cuilab.cn/sramp). D Western blot shoed the protein level of SLC7A11 in the cardiomyocytes (H9C2) under LPS treatment (10 µg/mL). E RIP-PCR analysis was performed to detect the molecular binding within SLC7A11 and METTL3. F MeRIP-PCR was performed to m6A modification level of SLC7A11 mRNA in cardiomyocytes under LPS treatment. Cellular experiments were performed in triplicate and were repeated three times. **p < 0.01
METTL3/YTHDF2 interacted with SLC7A11 mRNA
To discovery the potential bridge in the regulation of METTL3 to SLC7A11, several common m6A ‘reader’ were detected in the cardiomyocytes (H9C2) under LPS treatment (10 µg/mL). Results illustrated that YTHDF2 demonstrated a higher level upon LPS treatment (Fig. 4A). Moreover, for the molecular binding within SLC7A11 and YTHDF2, the RIP-PCR analysis was performed and results indicated that YTHDF2 significantly correlated to SLC7A11 (Fig. 4B). Besides, the interaction within SLC7A11 and YTHDF2 was degraded by the knockdown of METTTL3 (sh-METTL3) (Fig. 4C). Subsequently, the overexpression of YTHDF2 reduced the protein level of SLC7A11 (Fig. 4D). Overall, these data indicated that METTL3/YTHDF2 interacted with SLC7A11 mRNA.

METTL3/YTHDF2 interacted with SLC7A11 mRNA. A Several common classical m6A ‘reader’ were detected using RT-PCR in the cardiomyocytes (H9C2) under LPS treatment (10 µg/mL). B RIP-PCR analysis was performed to detect the molecular binding within SLC7A11 and YTHDF2 using anti-YTHDF2 antibody. C RIP-PCR analysis was performed to detect the binding within SLC7A11 and YTHDF2 upon METTTL3 knockdown (sh-METTL3) or controls (sh-NC). D Western blot analysis was performed to show the protein level of YTHDF2 and SLC7A11. Cellular experiments were performed in triplicate and were repeated three times. **p < 0.01; *p < 0.05
METTL3/YTHDF2 decayed the stability of SLC7A11 mRNA
Given the previous findings that YTHDF2 regulated the SLC7A11 expression, we further investigate the role of METTL3/YTHDF2 on SLC7A11 mRNA. Firstly, luciferase reporter assay using wild-type (WT) SLC7A11 3′UTR sequence or mutant (Mut) was performed (Fig. 5A). Results indicated that METTL3 silencing accelerated the luciferase activity within SLC7A11 wild type sequence. Results indicated that METTL3 modulated SLC7A11 expression mainly through the m6A site (Fig. 5B). RNA decay analysis revealed that METTL3 knockdown up-regulate the SLC7A11 mRNA stability, and YTHDF2 overexpression repressed the SLC7A11 mRNA stability (Fig. 5C). Moreover, the co-transfection of METTL3 knockdown and YTHDF2 overexpression recovered the stability of SLC7A11 that reduced by YTHDF2 overexpression (Fig. 5D). These results demonstrated that m6A writer METTL3 decayed the mRNA stability of SLC7A11 through YTHDF2/m6A/dependent manner.

METTL3/YTHDF2 enhanced the stability of SLC7A11 mRNA. A SLC7A11 mRNA 3’UTR containing m6A modification site was cloned into luciferase reporter vectors, including mutation (Mut) of m6A consensus sequence (GGACT) and mutant by replacing adenosine with cytosine. B The luciferase activity within SLC7A11 wild type sequence and METTL3 knockdown or control was detected. Relative luciferase activity was computed by the ratio of Firefly and Renilla luciferase values. C RNA decay analysis was performed to reveal the SLC7A11 mRNA remaining level under Act D treatment. Cells were transfected with METTL3 knockdown (sh-METTL3) or YTHDF2 overexpression (YTHDF2 OV), as well as controls. D RNA decay analysis was performed using the co-transfection of METTL3 knockdown and YTHDF2 overexpression. Cellular experiments were performed in triplicate and were repeated three times. **p < 0.01; *p < 0.05
METTL3/YTHDF2/SLC7A11 regulated the LPS-induced ferroptosis in cardiomyocytes
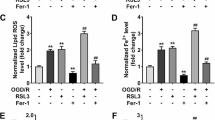
To identify the role of METTL3/YTHDF2/SLC7A11 axis on LPS-induced ferroptosis in cardiomyocytes, rescue assays were performed. Firstly, the SLC7A11 protein was repressed by the YTHDF2 overexpression and METTL3 silencing up-regulated it (Fig. 6A). For the death rate, results showed that SLC7A11 overexpression significantly decreased the death, and YTHDF2 overexpression up-regulated the death rate and METTL3 silencing repressed it (Fig. 6B). For the lipid ROS, results showed that SLC7A11 overexpression reduced the ROS level, and YTHDF2 overexpression increased the ROS and METTL3 silencing repressed it (Fig. 6C). For the iron load assay, results showed that SLC7A11 overexpression inhibited the Fe2+ concentration, and YTHDF2 overexpression enhanced it and METTL3 silencing repressed it (Fig. 6D). These results demonstrated that METTL3/YTHDF2/SLC7A11 regulated the LPS-induced ferroptosis in cardiomyocytes.

METTL3/YTHDF2/SLC7A11 regulated the LPS-induced ferroptosis in cardiomyocytes. A Western blot analysis was performed to detect the SLC7A11 protein level in the cardiomyocytes (H9C2) under LPS treatment (10 µg/mL). B The death rate was detected using flow cytometry in cardiomyocytes with SLC7A11 overexpression (SLC7A11 OV), SLC7A11 overexpression (SLC7A11 OE) and METTL3 silencing (sh-METTL3). C The lipid ROS was calculated using flow cytometry in cardiomyocytes with SLC7A11 overexpression (SLC7A11 OV), SLC7A11 overexpression (SLC7A11 OE) and METTL3 silencing (sh-METTL3). D GSH/GSSG was calculated in cardiomyocytes with SLC7A11 overexpression (SLC7A11 OV), SLC7A11 overexpression (SLC7A11 OE) and METTL3 silencing (sh-METTL3). E Iron load assay was performed in cardiomyocytes with SLC7A11 overexpression (SLC7A11 OV), SLC7A11 overexpression (SLC7A11 OE) and METTL3 silencing (sh-METTL3). Cellular experiments were performed in triplicate and were repeated three times. **p < 0.01; *p < 0.05
Discussion
In the intensive care unit, sepsis always acts as a common critical illness and causes major challenge in critical care medicine management and treatment. Sepsis is a life-threatening disorder, which results from infectious host response to dysfunction. Due to the rising incidence, sepsis exposure is emerging as a global burden, causing big challenge to intensive care clinician.
In ferroptosis progression, iron is involved in sepsis occurrence and development and iron chelators could inhibit the development of sepsis. The ferroptotic cells could release damage-associated molecular patterns and lipid peroxidation, which further mediates inflammatory responses [20]. Here, we found that the ferroptotic phenotypic characteristic showed a significant up-regulation in the sepsis-induced cardiomyocytes. Besides, the m6A modification level and m6A writer METTL3 protein both up-regulated in the LPS treatment. Thus, we put forward the hypothesis that METTL3 and m6A modification might regulate the progression of septic myocardial injury.
In the septic myocardial injury, our previous research had indicated that METTL3 and m6A modification level up-regulated in the lipopolysaccharide (LPS)-induced cardiomyocytes (H9C2 cells) Functionally, METTL3 knockdown inhibits the LPS-induced cardiomyocytes’ inflammatory damage. Mechanistically, there is a remarkable m6A modification site on HDAC4 mRNA 3′-UTR genome, and m6A reader IGF2BP1 recognized the m6A modification sites on HDAC4 mRNA and enhanced its mRNA stability. Thus, our previous research revealed a novel insight for METTL3 in the sepsis-induced myocardial injury. Here, in this present new study, we found that METTL3 could regulate the ferroptosis in the sepsis-induced myocardial injury, which is completely different from our previous direction, namely cardiomyocytes’ inflammatory damage. Therefore, this research expands our understanding of the function of METTL3.
Given that SLC7A11 acted as the downstream effector for METTL3/YTHDF2/m6A/SLC7A11 axis, the bio-function of SLC7A11 is of the essence. Several studies have shown that SLC7A11 is one of the key regulators of ferroptosis. The activation of antioxidant stress-related pathways, including the GSH system, plays an important role in sepsis treatment. SLC7A11 provides a substrate for the synthesis of GSH and is mainly involved in the regulation of redox status, ferroptosis and intercellular signaling. A critical finding inspired us that there is remarkable m6A modification in the SLC7A11 mRNA [16], thus we explore the function and interaction within METTL3 and SLC7A11. We found that there was a suitable m6A modified site was ‘GGACT’ in the 3′-UTR of SLC7A11 genome. Subsequently, the RIP-PCR and MeRIP-PCR assays were all performed and data suggested the m6A modification in SLC7A11.
YTHDF2 is a special m6A reader that mediates an RNA degradation in post-transcriptional regulation [21, 22]. Numerous findings revealed that YTHDF2 decays its target mRNA to regulate pathophysiological process. For example, in prostate cancer, YTHDF2 directly binds to the m6A modification sites of NKX3-1 and LHPP to mediate the mRNA degradation, and knock-down of YTHDF2 significantly induces NKX3-1 and LHPP expression at both mRNA and protein level with repressed phosphorylated AKT [23]. Lipid ROS often occurs in the process of ferroptosis, and the expression of anti-iron death marker GPX4 and SLC7A11 is down-regulated [24, 25]. Here, in present research, we found that SLC7A11 was down-regulated in the LPS treatment. Moreover, YTHDF2 directly bound to the SLC7A11 mRNA and significantly decay its mRNA stability. Thus, YTHDF2 exerted a negative regulation towards SLC7A11.
In summary, our work provided novel evidence to elucidate that METTL3 mediates the ferroptosis in septic myocardial injury. YTHDF2 mediates the mRNA degradation of the ferroptosis suppressor SLC7A11 in m6A-depnedent manner to regulate SLC7A11-regulated myocardial injury in sepsis (Fig. 7). We hope this study could provide a novel regulatory mechanism that may assist the development of potential diagnosis or therapeutic for sepsis in the future. In conclusion, the findings illustrated a role of METTL3/YTHDF2/m6A/SLC7A11 axis on sepsis-induced myocardial injury.

METTL3/YTHDF2/m6A/SLC7A11 axis regulates the septic myocardial injury regarding ferroptosis
Data Availability
No research data shared.
References
Boehm D, Menke H (2021) Sepsis in burns-lessons learnt from developments in the management of septic shock. Medicina (Kaunas, Lithuania) 58(1):26
Gandhirajan A, Roychowdhury S, Vachharajani V (2021) Sirtuins and sepsis: cross talk between redox and epigenetic pathways. Antioxidants (Basel, Switzerland) 11(1):3
Li Y, Zhao H, Guo Y, Duan Y, Guo Y, Ding X (2021) Association of preadmission metformin use and prognosis in patients with sepsis and diabetes mellitus: a systematic review and meta-analysis. Front Endocrinol 12:811776
Lin J, Tan B, Li Y, Feng H, Chen Y (2021) Sepsis-exacerbated brain dysfunction after intracerebral hemorrhage. Front Cell Neurosci 15:819182
Lopes-Pires ME, Frade-Guanaes JO, Quinlan GJ (2021) Clotting dysfunction in sepsis: a role for ROS and potential for therapeutic intervention. Antioxidants (Basel, Switzerland) 11(1):88
Guo L, Yang H, Zhou C, Shi Y, Huang L, Zhang J (2021) N6-methyladenosine RNA modification in the tumor immune microenvironment: novel implications for immunotherapy. Front Immunol 12:773570
Lu S, Ding X, Wang Y, Hu X, Sun T, Wei M et al (2021) The relationship between the network of non-coding RNAs-molecular targets and N6-methyladenosine modification in colorectal cancer. Front cell Dev Biol 9:772542
Liu Y, Song R, Zhao L, Lu Z, Li Y, Zhan X et al (2022) M(6)a demethylase ALKBH5 is required for antibacterial innate defense by intrinsic motivation of neutrophil migration. Signal Transduct Target therapy 7(1):194
Shen H, Xie K, Li M, Yang Q, Wang X (2022) N(6)-methyladenosine (m(6)A) methyltransferase METTL3 regulates sepsis-induced myocardial injury through IGF2BP1/HDAC4 dependent manner. Cell death discovery 8(1):322
Zhang S, Guan X, Liu W, Zhu Z, Jin H, Zhu Y et al (2022) YTHDF1 alleviates sepsis by upregulating WWP1 to induce NLRP3 ubiquitination and inhibit caspase-1-dependent pyroptosis. Cell Death Discov 8(1):244
Wang N, Yao F, Liu D, Jiang H, Xia X, Xiong S (2022) RNA N6-methyladenosine in nonocular and ocular disease. J Cell Physiol 237(3):1686–1710
Xu Y, Zhang M, Zhang Q, Yu X, Sun Z, He Y et al (2021) Role of main RNA methylation in hepatocellular carcinoma: N6-methyladenosine, 5-methylcytosine, and N1-methyladenosine. Front cell Dev Biol 9:767668
Li D, Pi W, Sun Z, Liu X, Jiang J (2022) Ferroptosis and its role in cardiomyopathy. Biomed Pharmacother 153:113279
Chen Z, Cao Z, Gui F, Zhang M, Wu X, Peng H et al (2022) TMEM43 protects against sepsis-induced cardiac injury via inhibiting ferroptosis in mice. Cells 11:19
Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR et al (2019) Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572(7769):402–406
Ji FH, Fu XH, Li GQ, He Q, Qiu XG (2022) FTO prevents thyroid cancer progression by SLC7A11 m6A methylation in a ferroptosis-dependent manner. Front Endocrinol 13:857765
Li XC, Jin F, Wang BY, Yin XJ, Hong W, Tian FJ (2019) The m6A demethylase ALKBH5 controls trophoblast invasion at the maternal-fetal interface by regulating the stability of CYR61 mRNA. Theranostics 9(13):3853–3865
Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M et al (2019) METTL3 and ALKBH5 oppositely regulate m(6)a modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 15(8):1419–1437
Li B, Zhu L, Lu C, Wang C, Wang H, Jin H et al (2021) circNDUFB2 inhibits non-small cell lung cancer progression via destabilizing IGF2BPs and activating anti-tumor immunity. Nat Commun 12(1):295
Liu Y, Tan S, Wu Y, Tan S (2022) The emerging role of ferroptosis in Sepsis. DNA Cell Biol 41(4):368–380
Zhang MM, Lin YL, Zeng WF, Li Y, Yang Y, Liu M et al (2021) N6-methyladenosine regulator-mediated immune genes identify breast cancer immune subtypes and predict immunotherapy efficacy. Front Genet 12:790888
Zhang N, Zuo Y, Peng Y, Zuo L (2021) Function of N6-methyladenosine modification in tumors. J Oncol 2021:6461552
Li J, Xie H, Ying Y, Chen H, Yan H, He L et al (2020) YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6-methyladenosine-dependent way in prostate cancer. Mol Cancer 19(1):152
Su J, Bian C, Zheng Z, Wang H, Meng L, Xin Y et al (2022) Cooperation effects of radiation and ferroptosis on tumor suppression and radiation injury. Front cell Dev biology 10:951116
Wang X, Wang Y, Huang D, Shi S, Pei C, Wu Y et al (2022) Astragaloside IV regulates the ferroptosis signaling pathway via the Nrf2/SLC7A11/GPX4 axis to inhibit PM2.5-mediated lung injury in mice. Int Immunopharmacol 112:109186
Acknowledgements
No.
Funding
This work was supported by National Natural Science Foundation of China (81870207).
Author information
Authors and Affiliations
Contributions
HS, KX and XW wrote the main manuscript text and prepared Figures 1–7. YT help improve the language. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shen, H., Xie, K., Tian, Y. et al. N6-methyladenosine writer METTL3 accelerates the sepsis-induced myocardial injury by regulating m6A-dependent ferroptosis. Apoptosis 28, 514–524 (2023). https://doi.org/10.1007/s10495-022-01808-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-022-01808-y