
Abstract
Myocardial damage is responsible for the high mortality of sepsis. However, the underlying mechanism is not well understood. Cardiomyocyte autophagy alleviates the cardiac injury caused by myocardial infarction. Enhanced cardiomyocyte autophagy also has protective effects against cardiomyocyte mitochondrial injury. Minocycline enhances autophagy in many types of cells under different types of pathological stress and can be easily taken up by cardiomyocytes. The present study investigated whether minocycline prevented myocardial injury caused by sepsis and whether cardiomyocyte autophagy participated in this process. The results indicated that minocycline enhanced cardiomyocyte mitochondrial autophagy and cardiomyocyte autophagy and improved myocardial mitochondrial and cardiac function. Minocycline upregulated protein kinase B (Akt) phosphorylation, inhibited mTORC1 expression and enhanced mTORC2 expression. In conclusion, minocycline enhanced cardiomyocyte mitochondrial autophagy and cardiomyocyte autophagy and improved cardiac function. The underlying mechanisms were associated with mTORC1 inhibition and mTORC2 activation. Thus, our findings suggest that minocycline may represent a potential approach for treating myocardial injury and provide novel insights into the underlying mechanisms of myocardial injury and dysfunction after sepsis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sepsis is a complex systemic disease that is defined as life-threatening organ dysfunction caused by the dysregulation of the host response to infection [1]. In China, intensive care unit (ICU) and hospital-wide mortality for sepsis are 28.7% and 33.5%, respectively [2]. Greater than 60% of sepsis patients admitted to the ICU present with cardiac dysfunction; the mortality rate for these patients ranges from 70 to 90% [3].
Myocardial injury is independently associated with early mortality and postdischarge cardiovascular morbidity in sepsis [4]. Sepsis-induced cardiomyocyte mitochondrial oxidative stress is major factor in myocardial injury [5]. Nonetheless, sepsis induces cardiomyocyte mitochondrial swelling, iliac crest loss, vacuole formation, declining membrane potential and other structural and functional changes, which lead to myocardial dysfunction [6]. However, how to improve myocardial injury by alleviating mitochondrial injury in sepsis remains unclear.
A previous study demonstrated that increased cardiomyocyte mitochondrial autophagy is beneficial for improving cardiomyocyte mitochondrial dysfunction and mitigating myocardial ischemia caused by heart failure [7]. Furthermore, minocycline is an inducer of autophagy in various pathological conditions, inhibits apoptosis in neuronal cells subjected to radiation [8] and exhibits protective effects in endothelial cells subjected to ischemia/reperfusion [9]. In addition, minocycline is easily taken up by myocardial tissue and significantly reduces myocardial infarct sizes (33%) by exerting antioxidation effects [10]. Thus, we speculate that minocycline maybe exert protective effects against sepsis-induced myocardial injury via inducing myocardial autophagy.
Mammalian target of rapamycin (mTOR) is an autophagy effector, and mTOR coordinates cell growth and plays a fundamental role in cell and organismal physiology [11]. Akt/mTOR is responsible for minocycline-mediated effects in ovarian tumors [12]. However, there are no reports of whether Akt/mTOR signaling is involved in the protective effects of minocycline in septic myocardial injury.
Thus, our goal was to understand whether Akt/mTOR signaling is the mechanism through which minocycline-induced cardiomyocyte mitochondrial autophagy protects against septic myocardial injury and improves cardiac function.
Methods
Animals and treatment
The Fourth Military Medical University Ethics Committee on Animal Care approved all animal protocols, and all experiments were performed in accordance with the National Institutes of Health Guidelines on the Use of Laboratory Animals. Male C57BL/6 of 8–12 weeks mice were obtained from the Experimental Animal Center of the Fourth Military Medical University. Polymicrobial sepsis was induced in anesthetized mice by cecal ligation and puncture (CLP) according to our previous protocol [13]. The detailed animal model construction method was included in the Supplementary Materials.
Experimental protocols
Mice were induced by CLP/sham and then were randomly divided into the predetermined groups according to different treatments, and the number of mice was determined according to different assessment indicators with predetermined time. The schematic diagram about the protocols was shown in Supplementary Fig. 1.
Neonatal cardiomyocyte culture
Primary cardiomyocyte cultures were harvested from the ventricles of neonatal Sprague Dawley (SD) rats (1 day old). Cells were cultured in DMEM supplemented with 15% fetal bovine serum (Gibco, CA, USA) and maintained at 37 °C in 95% O2 and 5% CO2 as previously described [14]. The detailed construction method was included in the Supplementary Materials.
LPS treatment
LPS (Sigma) was added to primary cardiomyocytes in serum-containing media for 24 h; then, minocycline and 3-methyladenine (3MA) treatments were administered.
Minocycline and 3-methyladenine treatment
The mice were treated with intraperitoneal injections of minocycline (Abcam) at 1 h, 25 h, and 49 h after CLP, and 3MA (Sigma-Aldrich), an autophagy inhibitor [15], was administered 30 min before every minocycline treatment time point. The control group received intraperitoneal injections of equal volumes of normal saline (NS). In vitro, primary cardiomyocytes were treated with minocycline (80 µg/ml) 1 h after LPS challenge, and 3MA (40 µm), was added 30 min before minocycline treatment. Equal volumes of sterile phosphate-buffered solution (PBS) were used as control.
Survival study
For the survival study, the CLP/sham-induced mice were returned to their cages after treatment administration and were closely monitored for up to 7 days, as well as given ad libitum access to food and water.
Determination of myocardium or cardiomyocyte apoptosis
Myocardium or cardiomyocyte apoptosis was determined by terminal deoxyribonucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining according to our previous study [16]. The detailed method was included in the Supplementary Materials.
Echocardiography
The CLP/sham-induced mice were sedated by 3% isoflurane inhalation and then assessed by echocardiography (Sequoia Acuson, 15-MHz linear transducer; Siemens, Erlangen, Germany). LV end-systolic volume (LVESV) and LV end-diastolic volume (LVEDV) were measured on the LV short axis. LV ejection fraction (LVEF) and LV fraction shortening (LVFS) were calculated using computer algorithms.
Transmission electron microscopy (TEM)
Autophagosomes and mitochondrial ultrastructure were detected by TEM as previously described [17]. The detailed method was included in the Supplementary Materials.
Determination of cTnI, CK-MB and LDH
At the predetermined time points, animals were anesthetized by isoflurane, and blood samples were collected from the mouse carotid artery and clotted for 30 min at 25 °C. The serum was separated by centrifugation at 2000 g for 15 min at 4 °C, aliquoted, and stored at − 80 °C until use in various biochemical assays. The concentrations of cTnI and CK-MB were measured using ELISA kits according to the manufacturer’s instructions (Beyotime Institute Biotechnology, Jiangsu Province, China). Cell death was assessed by the release of lactate dehydrogenase (LDH), a cytoplasmic enzyme and a marker of membrane integrity. After treatment, the samples were centrifuged at 10,000 g for 10 min, and the supernatants were collected for analysis using an LDH assay kit (Beyotime, Institute Biotechnology, Jiang Su Province, China).
Determination of Mn-SOD, ATP and citrate synthase enzyme activity
Manganese superoxide dismutase (Mn-SOD) was assayed by the Superoxide Dismutase Activity Assay kit (Abcam, ab65354) as previously described by Vives-Bauza [18]. Citrate synthase (CS) was measured using a commercially available CS activity assay kit (Sigma-Aldrich), and the adenosine triphosphate (ATP) content was measured using an ATP bioluminescent assay kit (Sigma-Aldrich) according to the standard protocols described in our previous study [19].
Western blot evaluation
Protein was extracted from myocardium tissues or cultured cardiomyocytes according to standard Invitrogen protocols (Invitrogen, Carlsbad, CA, USA) as previously described [14]. Total protein from myocardium tissues and cardiomyocytes was separated by SDS–PAGE, blotted and probed with the following antibodies: anti-Akt (Cell Signaling, Danvers, MA, USA); anti-phospho-Akt (Ser473; Cell Signaling); anti-mTOR (ab2732, Abcam); anti-phospho-mTOR (Ser2448, ab109268, Abcam); anti-raptor (ab40768, Abcam); anti-rictor (ab104838, Abcam); anti-β-actin (Santa Cruz, CA, USA); anti-GAPDH (ab9485, Abcam); anti-LC3A/B (ab128025, Abcam); anti-p62 (ab91526, Abcam); and anti-caspase-3 (Sigma-Aldrich). Bradford assays (Bio-Rad Laboratories, Hercules, CA, USA) were used to quantify the protein concentrations. The blots were visualized with a chemiluminescence system (Amersham Bioscience, Buckinghamshire, UK), and the signals were quantified by densitometry.
Statistical analysis
The measurement data are expressed as the mean ± S.E.M. Comparisons between groups were determined by ANOVA followed by Bonferroni correction for post hoc t-tests. The data expressed as proportions were assessed using the Chi square test. Two-sided tests were used in this analysis. The survival studies were analyzed using the log-rank test. The survival rates are expressed as percentages. To demonstrate that 3MA could abolish minocycline-induced autophagy, we used Student’s t test to compare the differences between two groups. P < 0.05 was considered significant. SPSS software package version 14.0 (SPSS, Chicago, IL, USA) was used for the data analyses.
Results
Minocycline improves survival and cardiac function after sepsis
To investigate the impact of minocycline exposure on myocardial injury and cardiac dysfunction caused by sepsis, C57BL/6 mice were subjected to CLP. Then, mice were treated with minocycline for three consecutive days after CLP. First, we treated C57BL/6 septic mice with 25 mg/kg, 50 mg/kg or 100 mg/kg minocycline. Then, myocardial injury was assessed by serum cTnI levels, and survival was determined. The results revealed a dose-dependent reduction of cTnI in the CLP + Min group compared with the CLP + NS group (Fig. 1a). Only the 50 mg/kg and 100 mg/kg doses significantly reduced cTnI levels in CLP-induced mice, whereas cTnI levels were significantly increased by CLP compared with the Sham + NS group (Fig. 1a). Consistently, the survival was also dose dependently increased in the CLP + Min group compared with the CLP + NS group, and the significant beneficial effects of minocycline were noted with the 50 mg/kg and 100 mg/kg doses, whereas survival was significantly reduced by CLP compared with the Sham + NS group (Fig. 1b). Thus, we administered 50 mg/kg minocycline for the subsequent in vivo experiments. Next, to further investigate whether minocycline-mediated improvement in cardiac function facilitated improvement in survival, we used ultrasound technology to calculate the ejection fraction (EF) and fractional shortening (FS) (Fig. 1c). EF and FS were significantly increased in CLP + Min group compared with CLP + NS group, whereas EF and FS were significantly reduced by CLP compared with the Sham + NS group (Fig. 1d, e). These results indicated that minocycline might prevent cardiac dysfunction caused by CLP.

Minocycline improves survival and cardiac function after sepsis. Sepsis was induced by CLP. Dose-dependent effects on myocardial injury and survival after minocycline treatment was assessed Treatments were performed on animals by the intraperitoneal injection of minocycline at 1 h, 25 h, and 49 h after surgery. a cTnI (n = 6). b 7-days survival, the values are expressed as surviving percentage (n = 20). c On the third day after surgery, heart function was evaluated by ultrasound and echocardiography (n = 8). d and e Statistical analysis of EF and FS. The data are expressed as the mean ± S.E.M. **P < 0.01, ***P < 0.001 versus Sham + NS; ##P < 0.01, ###P < 0.001 versus CLP + NS. Min-25: 25 mg/kg minocycline, Min-50: 50 mg/kg minocycline, Min-100: 100 mg/kg minocycline
Minocycline induces myocardial autophagy to prevent myocardial apoptosis and injury
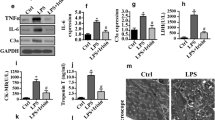
To preliminarily explore whether minocycline-induced myocardial autophagy exerted protection effects on myocardial injury, we obtained a homogenate of myocardial tissue 24 h after minocycline or NS treatment in CLP/sham-induced mice. The autophagy-related proteins LC3 and p62 were assessed by western blot (Supplementary Fig. 2a). The results showed that minocycline treatment significantly increased the LC3II/LC3I ratio and decreased p62 compared with NS treatment in septic mice (Supplementary Fig. 2b, c). This finding indicated that minocycline might induce myocardial autophagy in septic mice. Next, to further investigate whether minocycline-induced myocardial autophagy exerted protection effects on myocardial injury, we used the proven autophagy inhibitor 3MA (dose dependent: 30 mg/kg or 60 mg/kg) to treat CLP-induced mice 30 min before minocycline administration. The result suggested that 60 mg/kg 3MA significantly reversed the survival improvement induced by minocycline (Supplementary Fig. 3a). Moreover, 60 mg/kg 3MA also reversed the minocycline-mediated reduction in cTnI levels (Supplementary Fig. 3b). Thus, we administered 60 mg/kg 3MA for the subsequent in vivo experiments. Next, levels of the myocardial autophagy-related proteins LC3 and p62 in homogenized myocardial tissues were measured by western blot (Fig. 2a). The results showed that the LC3II/LC3I ratio was lower in the CLP + NS group than in the Sham + NS group; however, the LC3II/LC3I ratio was significantly higher in the CLP + Min group than in the CLP + NS group (Fig. 2b). Conversely, p62 expression was significantly higher in the CLP + NS group than in the Sham + NS group, but lower in the CLP + Min group than in the CLP + NS group (Fig. 2c). Interestingly, the minocycline-mediated increases in the LC3II/LC3I ratio and decreases in the p62 expression were significantly reversed by 3MA (Fig. 2b, c). Next, we observed CLP-induced myocardial apoptosis after minocycline administration with or without 3MA by TUNEL staining (Fig. 2d). The results revealed that compared with NS treatment, minocycline inhibited myocardial apoptosis caused by CLP and significantly reduced the apoptosis index, whereas myocardial apoptosis was higher in the CLP group than in the sham group (Fig. 2d, e). However, compared with CLP + Min group, 3MA abolished the minocycline-mediated prevention myocardial apoptosis, and the apoptosis index significantly increased (Fig. 2e). Consistently, minocycline inhibited caspase-3 expression induced by CLP, and caspase-3 expression was higher in the CLP + NS group than in the Sham + NS group (Fig. 2f). 3MA abolished the minocycline-mediated inhibition of caspase 3 expression (Fig. 2f). Finally, we assessed myocardial injury by measuring serum cTnI and CK-MB levels. The results showed that cTnI and CK-MB levels were higher in the CLP + NS group than in the Sham + NS group, but lower in the CLP + Min group than in the CLP + NS group (Fig. 2g, h). Moreover, the minocycline-mediated reductions in cTnI and CK-MB levels were also significantly reversed by 3MA (Fig. 2g, h). Thus, we infer that minocycline may prevent myocardial apoptosis and injury through inducing myocardial autophagy.

Minocycline induces myocardial autophagy to prevent myocardial apoptosis and injury. Sepsis was induced by CLP, and 50 mg/kg of minocycline was administered by intraperitoneal injection. One hour before minocycline treatment, 3MA was administered by intraperitoneal injection at predetermined time points. a Detection LC3 and p62 expression by western blot in myocardial homogenates. b and c Statistical analysis of the LC3II/LC3I ratio and p62 relative to GAPDH. Myocardial injury was evaluated based on the myocardial apoptosis index, which is the percentage of TUNEL-positive cells over the total nuclei as determined by DAPI staining from a total of 50 fields per heart sample (bar = 100 µm, n = 6) (d) and (e). f Representative gel blots and quantification of protein expression of caspase 3. g and h Evaluation of myocardial injury by cTnI and CK-MB in serum (n = 6). The data are expressed as the mean ± S.E.M.*P < 0.05, **P < 0.01, ***P < 0.001 versus Sham + NS; #P < 0.05, ##P < 0.01, ###P < 0.001 versus CLP + NS; $P < 0.05, $$P < 0.01, $$$P < 0.001 versus CLP + Min
Minocycline improves myocardial mitochondrial function and injury by inducing myocardial mitochondrial autophagy
Mitochondria are the source of energy for cells survival. Mitochondrial autophagy has been suggested to contribute to the maintenance of mitochondrial quality and quantity [20]. Thus, we explored whether minocycline induced myocardial mitochondrial autophagy and thus exerted beneficial effects on improving mitochondrial function. We used TEM to assess the myocardial ultrastructure. Our results indicated that compared with NS treatment, minocycline significantly increased the number of myocardial mitochondrial autophagosomes (indicated by the red arrows) in septic mice, whereas the number of myocardial mitochondrial autophagosomes was reduced in the CLP + NS group compared with the sham + NS group (Fig. 3a, b). However, 3MA reversed the minocycline-mediated increase in myocardial mitochondrial autophagosome (Fig. 3a, b). We also assessed the myocardial mitochondrial ultrastructure by TEM, and the results revealed that CLP induced the disorganized, damaged mitochondrial architecture in myocardium (mitochondrial swelling, iliac crest loss and vacuole formation) (Fig. 3c). In contrast, minocycline alleviated the impairments in myocardial mitochondrial ultrastructure (Fig. 3c). As expected, 3MA abolished the minocycline-mediated protection against myocardial mitochondrial injuries (Fig. 3c). Finally, we assessed myocardial mitochondrial function according to Mn-SOD, ATP and CS enzyme activity. The results showed that ATP levels and CS enzyme activity were reduced, and Mn-SOD levels were increased in the CLP + NS group compared with the Sham + NS group, whereas minocycline significantly improved ATP levels and CS enzyme activity and decreased Mn-SOD levels in the CLP-induced mice (Fig. 3d–f). However, 3MA inhibited the minocycline-mediated mitochondrial function improvements by reducing the ATP levels and CS enzyme activity and by increasing Mn-SOD levels (Fig. 3d–f). We infer that minocycline-induced myocardial mitophagy protected against mitochondrial ultrastructure impairment and thus improved myocardial mitochondrial function.

Minocycline improves myocardial mitochondrial function and reduces myocardial mitochondrial injury by inducing autophagy in myocardial mitochondria. Samples were obtained 24 h after minocycline treatment. a Myocardial mitochondrial autophagy was evaluated by TEM (indicated by the red box and red arrows). b The number of mitochondrial autophagosomes in myocardium was calculated per field from 10 fields per sample (bar = 2 µm, n = 6). c Myocardial mitochondrial ultrastructure changes evaluated by TEM (indicated by the red box and the red arrows). CLP induced mitochondrial swelling, iliac crest loss, vacuole formation and other structural changes in the myocardium; minocycline prevented these changes in the myocardial mitochondria, and 3MA reversed the minocycline-induced improvements. d, e and f Myocardial mitochondrial function was evaluated by ATP, CS enzyme activity and Mn SOD (n = 6). The data are expressed as the mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001 versus Sham + NS; #P < 0.05, ##P < 0.01, ###P < 0.001 versus CLP + NS; $P < 0.05, $$$P < 0.001 versus CLP + Min
Minocycline improves survival by improving cardiac function through myocardial autophagy induction
We further explored whether the minocycline-mediated improvements in cardiac function through myocardial autophagy. We used echocardiography to assess EF, ES, LVESV and LVEDV (Fig. 4a). Statistical analysis revealed that EF and FS were significantly reduced and that LVESV was significantly increased in the CLP + NS group compared with the Sham + NS group (Fig. 4b–d). However EF and FS were significantly improved and LVESV was significantly reduced in the CLP + Min group compared with the CLP + NS group (Fig. 4b–d). Interestingly, minocycline-mediated improvements in EF, FS and LVESV were significantly reversed by 3MA (Fig. 4b–d). However, LVEDV showed no significant differences between the groups (Fig. 4e). The mouse weights were also not significantly different between the groups (Fig. 4f). Heart function improvement is beneficial in sepsis; therefore, we also assessed the survival. The results demonstrated that minocycline significantly improved the survival of septic mice (Fig. 4g). This minocycline-mediated improvement in the survival was reversed by 3MA (Fig. 4g). This result was consistent with previous results (Supplementary Fig. 3a). Together, these results indicated that minocycline improves survival in septic mice by improving cardiac function through the induction of myocardial autophagy or myocardial mitochondrial autophagy.

Minocycline induces myocardial autophagy to improve cardiac function and survival. a Twenty-four hours after the end of minocycline treatment, heart function was evaluated by echocardiography. b, c, d and e Statistical analysis of EF, FS, LVESV and LVEDV (n = 8). f Mouse weights were assessed (n = 8 for each group). The data are expressed as the mean ± S.E.M. g 7-days survival, the values are expressed as surviving percentage (n = 20). **P < 0.01, ***P < 0.001 versus Sham + NS; #P < 0.05, ##P < 0.01, ###P < 0.001 versus CLP + NS; $P < 0.05, $$P < 0.01, $$$P < 0.001 versus CLP + Min
Minocycline induces cardiomyocyte autophagy to prevent cardiomyocyte apoptosis caused by LPS
To investigate the impact of minocycline on cardiomyocyte autophagy in vitro, primary cardiomyocytes were stimulated with various doses of LPS (0.1 µg/ml, 1 µg/ml and 10 µg/ml) The apoptosis and LDH levels were assessed to determine the effects of LPS on cardiomyocyte injury. The results showed that cardiomyocyte apoptosis and LDH levels were significantly increased in parallel with the increasing LPS dose (Supplementary Fig. 4a, b). Thus, we treated primary cardiomyocytes with 5 µg/ml LPS for all subsequent in vitro experiments. Next, TEM revealed that the number of autophagosomes (indicated by red arrows, Fig. 5a) was significantly reduced in the LPS-treatment group compared with the control group (Fig. 5b), whereas the number of autophagosomes was significantly increased upon minocycline in LPS-induced cardiomyocytes (Fig. 5b). Moreover, 3MA abolished the minocycline-mediated increase in the number of autophagosomes (Fig. 5b). We also assessed mitochondrial structure. The results revealed that LPS induced the disorganized, damaged ultrastructure of cardiomyocytes (mitochondrial swelling, iliac crest loss, volume diminished) (Supplementary Fig. 5). In contrast, compared with LPS + PBS group, minocycline alleviated the mitochondrial ultrastructure impairment caused by LPS (Supplementary Fig. 5). As expected, 3MA abolished minocycline-mediated protection against mitochondrial injuries in LPS-induced cardiomyocytes (Supplementary Fig. 5). Damage to the mitochondrial structure reduces mitochondrial function; therefore, we evaluated mitochondrial function based on ATP and CS enzyme activity assessments. The results showed that ATP levels and CS enzymatic activity were obviously decreased in the LPS + PBS group compared with the control group (Fig. 5c, d), whereas minocycline significantly improved ATP levels and CS enzymatic activity in LPS-induced cardiomyocytes (Fig. 5c, d). However, 3MA significantly inhibited the minocycline-mediated increase in mitochondrial function (Fig. 5c, d). We also assessed LHD levels, and the results indicated that minocycline significantly reduced LDH levels compared with PBS in LPS-induced cardiomyocytes (Fig. 5e). In addition, 3MA abolished the minocycline-mediated LDH level reductions (Fig. 5e). Finally, we assessed cardiomyocyte apoptosis using TUNEL staining (Fig. 5f). The results revealed that the apoptosis index was increased in the LPS + PBS group compared with the control group (Fig. 5f). Compared with the LPS + PBS group, minocycline significantly reduced the apoptosis index (Fig. 5f). However, 3MA abolished the protective effects of minocycline on cardiomyocyte apoptosis (Fig. 5f). Consistently, minocycline inhibited LPS-induced caspase-3 expression (Fig. 5g), and 3MA abolished the minocycline-mediated inhibition of caspase-3 expression (Fig. 5g). We thus infer that minocycline may prevent cardiomyocyte apoptosis and cardiomyocyte mitochondrial damage derived from LPS by inducing cardiomyocyte autophagy.

Minocycline improves cardiomyocyte survival by inducing autophagy in cardiomyocytes induced by LPS. Sepsis was induced in primary cardiomyocytes using LPS. Cardiomyocyte samples were obtained 24 h after minocycline treatment. a Cardiomyocyte autophagy was evaluated by TEM, and the number of autophagosomes in cardiomyocytes was calculated per cell from 10 cells per sample (bar = 2 µm, n = 6) (b). c and d Cardiomyocyte mitochondrial function was evaluated by ATP and citrate synthase enzyme activity. e LDH. f Representative images of TUNEL-stained primary neonatal cardiomyocytes and apoptosis index are shown in the right panel (bar = 50 µm, 50 fields per sample, n = 6). Representative gel blots and quantification of protein expression of caspase 3 and β-actin (loading control) (g). The data are expressed as the mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; #P < 0.05, ##P < 0.01, ###P < 0.001 versus LPS + PBS; $P < 0.05, $$$P < 0.001 versus LPS + Min
Targeting the Akt/mTOR pathway with minocycline strongly upregulated cardiomyocyte autophagy in sepsis
mTOR is the master regulator of cell growth and a vital molecule for inducing cell autophagy [11, 21]. To explore the impact of minocycline on mTOR phosphorylation and the related mechanism in cardiomyocyte autophagy in sepsis, we assessed the expression of autophagy-related proteins by western blot in LPS-induced primary cardiomyocytes (Fig. 6a). Western blot analyses demonstrated that the Akt/GAPDH ratio was not significantly different between the groups (Fig. 6b). However, the activated Akt (phosphorylated Akt, p-Akt)/GAPDH ratio was significantly reduced in the LPS + PBS group compared with the control group and was significantly increased in the LPS + Min group compared with the LPS + PBS group (Fig. 6c). Previous studies have indicated that p-Akt inhibits mTOR activation (phosphorylated mTOR, p-mTOR) [22]. Consistently, our results revealed that there were no significant differences in the mTOR/GAPDH ratio between the groups (Fig. 6d). However, p-mTOR was significantly reduced in the LPS + Min group compared with the LPS + PBS group, and p-mTOR was significantly increased in the LPS + PBS group compared with the control group (Fig. 6e). Moreover, mTORC1, which was identified by the association of mTOR with rapamycin-associated protein of TOR (raptor) [21] and was shown to inhibit autophagy [23], was reduced in the LPS + Min group compared with the LPS + PBS group, whereas raptor was significantly increased in the LPS + PBS group compared with the control group (Fig. 6f). mTORC2 is involved in other distinct signaling complexes and associates with rapamycin insensitive companion of mTOR (rictor) [21] and promotes autophagy [24]. Rictor levels were increased in the LPS + Min group compared with the LPS + PBS group, whereas rictor levels were significantly reduced in the LPS + PBS group compared with the control group (Fig. 6g). Interestingly, 3MA, which blocks Akt activation [25] abolished the minocycline-mediated increase in p-Akt levels (Fig. 6c). As expected, the reduction in p-mTOR and raptor levels after minocycline administration was abolished by 3MA (Fig. 6e, f). In contrast, 3MA abolished the minocycline-mediated increase in rictor levels (Fig. 6g). We thus infer that minocycline administration upregulated p-Akt and further inhibited p-mTOR and raptor activation; in contrast, minocycline promoted rictor activation. Next, we assessed the autophagic marker proteins LC3 and p62 by western blot with or without 3MA treatment (Fig. 6h) to further clarify the mechanism of minocycline-mediated autophagy in LPS-induced cardiomyocytes. The results showed that minocycline increased the LC3II/LC3I ratio and decreased p62 expression in LPS-induced cardiomyocytes (Fig. 6i, j). As expected, 3MA abolished the minocycline-mediated increase in the LC3II/LC3I ratio and decrease in p62 (Fig. 6i, j). All of these results indicated that minocycline administration inhibited mTORC1 by activating Akt and activated mTORC2 to further induce autophagy in LPS-induced primary cardiomyocytes.

Minocycline induces cardiomyocyte autophagy through Akt/mTOR signaling. Whole proteins from LPS-induced primary cardiomyocytes were obtained at 24 h after minocycline treatment with or without 3MA administration. a Representative blots and (b–g) analysis of Akt (b), p-Akt (c), mTOR (d), p-mTOR (e), raptor (f) and rictor (g). h Representative blots and analysis of LC3II/LC3I (i) and p62 (j) (n = 6). The data are expressed as the mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; #P < 0.05, ##P < 0.01, ###P < 0.001 versus LPS + PBS; $P < 0.05, $$P < 0.01, $$$P < 0.001 versus LPS + Min
Discussion
Sepsis is the world’s leading killer,five of the top 10 WHO causes of death fulfil the definition of sepsis (http://www.who.int). Sepsis-induced myocardial dysfunction is one of the major healthcare problems worldwide. Studies in animals showed that maintenance of mitochondrial integrity reduced post-cardiac arrest myocardial dysfunction and mortality [26]. Sepsis triggers myocardial mitochondrial structure damage and loss of mitochondrial density [27]. Currently, little is known regarding reducing myocardial mitochondrial damage to improve sepsis outcomes. The major findings from our current study revealed minocycline administration significantly induced myocardial mitochondrial autophagy to reduce mitochondrial damage, to prevent cardiomyocyte apoptosis, to improve cardiac function, and thus improved survival in CLP-induce mice. Moreover, minocycline administration significantly activated Akt, further inhibited p-mTOR and mTORC1 (autophagy inhibiting), significantly activated mTORC2 (autophagy inducing), and thus increased the autophagic LC3II/LC3I protein ratio and decreased p62 expression. These data indicate that minocycline induce cardiomyocyte mitochondrial autophagy to improve cardiac function and promote sepsis survival via Akt/mTOR-mediated mechanisms.
Mitochondrial dysfunction plays a significant role in the pathogenesis of sepsis, and the degree of mitochondrial dysfunction correlates with outcomes. Cardiomyocytes exhibit mitochondrial ultrastructural damage in both septic animals and patients [28]. Our results suggested that sepsis induced mitochondrial swelling, iliac crest loss and vacuole formation and further caused cardiomyocyte mitochondrial dysfunction by reducing ATP and CS enzyme activity and increasing Mn-SOD. Studies have demonstrated that mitochondrial dysfunction triggers apoptosis in cardiac myocytes [29]. Our results also suggested that sepsis induced cardiomyocytes apoptosis in CLP-induced mice and LPS-induced primary cardiomyocytes.
Mitochondrial autophagy is beneficial in improving mitochondrial damage [30], and a previous study demonstrated that rescuing mitochondrial autophagy attenuates mitochondrial dysfunction [7]. Although minocycline, an antibiotic drug, prevents nerve injury [31], studies have demonstrated the therapeutic potential of minocycline due to its modulation of autophagy and apoptosis [9, 32]. Our study demonstrated that minocycline significantly induced myocardial mitochondrial autophagy according to the increased number of mitochondrial autophagosomes in the myocardia in CLP-induced mice. Furthermore, minocycline also significantly induced cardiomyocyte autophagy in LPS-induced cardiomyocytes according to the increased number of autophagosomes in cardiomyocytes. Accompanying this effect, minocycline alleviated mitochondrial damage as indicated by the reversed mitochondrial ridge fusion and disappearance, further improved mitochondrial function as indicated by increased CS enzymatic activity and ATP production and by reduced Mn-SOD levels. Our study also demonstrated that minocycline administration significantly protected against myocardial cell apoptosis in septic mice and cardiomyocyte apoptosis in LPS-induced cardiomyocytes. These findings are consistent with a previous study reporting that inducing autophagy attenuates cardiomyocyte apoptosis induced by hypoxia [33].
Furthermore, 3MA reversed minocycline-induced myocardial mitochondrial autophagy by reducing the numbers of myocardial mitochondrial autophagosomes and cardiomyocyte autophagosomes accompanying reducing ATP content and CS enzyme activity and increasing Mn-SOD content. 3MA also reversed minocycline-induced improvements in cardiomyocyte apoptosis. These results suggested that minocycline may serve as an inducer of myocardial mitochondrial autophagy to protect against myocardial mitochondrial damage in septic mice, and thus prevent cardiomyocyte apoptosis in sepsis.
Cardiac dysfunction represents a clinical feature of sepsis and contributes to its mortality. Cardiomyocyte apoptosis is one of the major pathogenic mechanisms underlying myocardial injury and cardiac dysfunction caused by sepsis [34]. Our results suggested that CLP significantly reduced cardiac function represented by lower EF and FS and higher LVESV and significantly induced cardiomyocyte apoptosis, accompanied by increased mortality. Previous studies have suggested that reducing mitochondrial damage and increasing ATP production decrease myocardial cell apoptosis, which is essential for improving myocardial contractility [35]. Our study demonstrated that minocycline administration significantly increased ATP production and reduced myocardial apoptosis in septic mice, thus enhanced the myocardial contractile function according to the improved EF, FS and LVESV, accompanied by reduced mortality. However, 3MA abolished these improvements in septic mice. This finding is consistent with a previous study that a reduction in myocardial apoptosis by myocardial autophagy may be important for treating patients with heart dysfunction [36].
Although autophagy serves as a double-edged sword in the heart under conditions of stress [37], we found that the induction of myocardial mitochondrial autophagy by minocycline exerted protective effects on cardiac dysfunction subjected by sepsis.
Among the many kinases, PI3K/Akt is reported to play a pivotal role in the survival of cardiomyocytes [38]. As a phosphorylation kinase, Akt has greater than 30 downstream substrates [39], and activated Akt induces autophagy [21, 22]. In the present study, the antiapoptotic effect of minocycline, significantly activated Akt (p-Akt) in LPS-induced primary cardiomyocytes. Previous studies have demonstrated that p-Akt inhibits mTORC1 activation [40]. mTORC1 comprises three core components: mTOR, raptor, and mammalian lethal with Sec13 protein 8 (mLST8, also known as GßL). mTORC1 integrates nutrient and growth factor signaling to promote anabolic metabolism processes, such as protein synthesis and lipid synthesis, and inhibit catabolic pathways, such as lysosome biogenesis and autophagy [21]. Consistently, our results demonstrate that mTORC1 downregulation by p-Akt after minocycline administration significantly upregulated autophagy, which was indicated by an increase in the LC3II/LC3I ratio and decreased p62 expression. However, minocycline increased p-Akt, which was abolished by the 3MA in primary cardiomyocytes subjected to LPS. Furthermore, the minocycline-induced reduction in mTORC1 and p62 and increase in LC3II/LC3I were also abolished by 3MA.
Additionally, mTORC2 is another form of mTOR, which comprises mTOR, mLST8, and rictor and controls cell proliferation and survival, the activated mTORC2 independently phosphorylates Akt [21]. Interestingly, mTORC2 activation was significantly induced by minocycline, which is consistent with a previous study demonstrated that Akt activation is independently induced by mTORC2 activation [41]. Therefore, mTORC2 activation by minocycline promoted Akt activation and thus significantly upregulated cardiomyocyte autophagy in our present study. These findings indicate that minocycline induces autophagy partially by activating the mTORC2/Akt pathway.
In the present study, minocycline exerted its protective effects against sepsis-induced cardiomyocyte injury through Akt/mTOR signaling. However, whether the classical PI3K/Akt/mTOR signaling pathway induces autophagy is not investigated in our article, and how minocycline regulates mTORC2 signaling inducing autophagy needs to be further investigated.
In conclusion, the salient finding of the present study is that minocycline induces cardiomyocyte autophagy, particularly myocardial mitochondrial autophagy, and alleviates cardiac dysfunction caused by sepsis. These effects are accompanied by decreased cardiomyocyte apoptosis and improved myocardial mitochondrial function. Minocycline appears to induce cardiomyocyte and cardiomyocyte mitochondrial autophagy via an Akt/mTOR-dependent pathway.
References
Seymour CW, Liu VX, Iwashyna TJ et al (2016) Assessment of clinical criteria for sepsis: for the third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315(8):762–774
Zhou J, Qian C, Zhao M et al (2014) Epidemiology and outcome of severe sepsis and septic shock in intensive care units in mainland china. PLoS ONE 9(9):e107181
Vieillard-Baron A, Caille V, Charron C et al (2008) Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med 36(6):1701–1706
Frencken JF, Donker DW, Spitoni C et al (2018) Myocardial injury in patients with sepsis and its association with long-term outcome. Circ Cardiovasc Qual Outcomes 11(2):e004040
Han D, Li X, Li S et al (2017) Reduced silent information regulator 1 signaling exacerbates sepsis-induced myocardial injury and mitigates the protective effect of a liver X receptor agonist. Free Radic Biol Med 113:291–303
Martin L, Peters C, Heinbockel L et al (2016) The synthetic antimicrobial peptide 19–2.5 attenuates mitochondrial dysfunction in cardiomyocytes stimulated with human sepsis serum. Innate Immun 22(8):612–619
Shirakabe A, Zhai P, Ikeda Y et al (2016) Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 133(13):1249–1263
Zhang L, Huang P, Chen H et al (2017) The inhibitory effect of minocycline on radiation-induced neuronal apoptosis via AMPKalpha1 signaling-mediated autophagy. Sci Rep 7(1):16373
Dong W, Xiao S, Cheng M et al (2016) Minocycline induces protective autophagy in vascular endothelial cells exposed to an in vitro model of ischemia/reperfusion-induced injury. Biomed Rep 4(2):173–177
Romero-Perez D, Fricovsky E, Yamasaki KG et al (2008) Cardiac uptake of minocycline and mechanisms for in vivo cardioprotection. J Am Coll Cardiol 52(13):1086–1094
Boutouja F, Stiehm CM, Platta HW (2019) mTOR: a cellular regulator interface in health and disease. Cells 8(1):1–23
Ataie-Kachoie P, Pourgholami MH, Bahrami BF et al (2015) Minocycline attenuates hypoxia-inducible factor-1alpha expression correlated with modulation of p53 and AKT/mTOR/p70S6K/4E-BP1 pathway in ovarian cancer: in vitro and in vivo studies. Am J Cancer Res 5(2):575–588
Zhang EF, Hou ZX, Shao T et al (2017) Combined administration of a sedative dose sevoflurane and 60% oxygen reduces inflammatory responses to sepsis in animals and in human PMBCs. Am J Transl Res 9(6):3105–3119
Hu J, Man W, Shen M et al (2016) Luteolin alleviates post-infarction cardiac dysfunction by up-regulating autophagy through Mst1 inhibition. J Cell Mol Med 20(1):147–156
Cheng NT, Meng H, Ma LF et al (2017) Role of autophagy in the progression of osteoarthritis: the autophagy inhibitor, 3-methyladenine, aggravates the severity of experimental osteoarthritis. Int J Mol Med 39(5):1224–1232
Zhang M, Zhang L, Hu J et al (2016) MST1 coordinately regulates autophagy and apoptosis in diabetic cardiomyopathy in mice. Diabetologia 59(11):2435–2447
Wang T, Zhang L, Hu J et al (2016) Mst1 participates in the atherosclerosis progression through macrophage autophagy inhibition and macrophage apoptosis enhancement. J Mol Cell Cardiol 98:108–116
Vives-Bauza C, Starkov A, Garcia-Arumi E (2007) Measurements of the antioxidant enzyme activities of superoxide dismutase, catalase, and glutathione peroxidase. Methods Cell Biol 80:379–393
Sun D, Li S, Wu H et al (2015) Oncostatin M (OSM) protects against cardiac ischaemia/reperfusion injury in diabetic mice by regulating apoptosis, mitochondrial biogenesis and insulin sensitivity. J Cell Mol Med 19(6):1296–1307
Yamashita SI, Kanki T (2017) How autophagy eats large mitochondria: autophagosome formation coupled with mitochondrial fragmentation. Autophagy 13(5):980–981
Saxton RA, Sabatini DM (2017) mTOR Signaling in growth, metabolism, and disease. Cell 168(6):960–976
Chen D, Lin X, Zhang C et al (2018) Dual PI3K/mTOR inhibitor BEZ235 as a promising therapeutic strategy against paclitaxel-resistant gastric cancer via targeting PI3K/Akt/mTOR pathway. Cell Death Dis 9(2):123
Kundu M (2014) Too sweet for autophagy: hexokinase inhibition of mTORC1 activates autophagy. Mol Cell 53(4):517–518
Lampada A, O’Prey J, Szabadkai G et al (2017) mTORC1-independent autophagy regulates receptor tyrosine kinase phosphorylation in colorectal cancer cells via an mTORC2-mediated mechanism. Cell Death Differ 24(6):1045–1062
Lin YC, Kuo HC, Wang JS et al (2012) Regulation of inflammatory response by 3-methyladenine involves the coordinative actions on Akt and glycogen synthase kinase 3beta rather than autophagy. J Immunol 189(8):4154–4164
Huang CH, Tsai MS, Chiang CY et al (2015) Activation of mitochondrial STAT-3 and reduced mitochondria damage during hypothermia treatment for post-cardiac arrest myocardial dysfunction. Basic Res Cardiol 110(6):59
Doerrier C, Garcia JA, Volt H et al (2016) Permeabilized myocardial fibers as model to detect mitochondrial dysfunction during sepsis and melatonin effects without disruption of mitochondrial network. Mitochondrion 27:56–63
Soriano FG, Nogueira AC, Caldini EG et al (2006) Potential role of poly(adenosine 5′-diphosphate-ribose) polymerase activation in the pathogenesis of myocardial contractile dysfunction associated with human septic shock. Crit Care Med 34(4):1073–1079
Barile L, Lionetti V, Cervio E et al (2014) Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res 103(4):530–541
Thomas HE, Zhang Y, Stefely JA et al (2018) Mitochondrial complex I activity is required for maximal autophagy. Cell Rep 24(9):2404–2417.e2408
Liu Z, Liang Y, Wang H et al (2017) LncRNA expression in the spinal cord modulated by minocycline in a mouse model of spared nerve injury. J Pain Res 10:2503–2514
Liu WT, Lin CH, Hsiao M et al (2011) Minocycline inhibits the growth of glioma by inducing autophagy. Autophagy 7(2):166–175
Zhang J, He Z, Xiao W et al (2016) Overexpression of BAG3 attenuates hypoxia-induced cardiomyocyte apoptosis by inducing autophagy. Cell Physiol Biochem 39(2):491–500
Wang L, Li Y, Ning N et al (2018) Decreased autophagy induced by beta1-adrenoceptor autoantibodies contributes to cardiomyocyte apoptosis. Cell Death Dis 9(3):406
Guichard JL, Rogowski M, Agnetti G et al (2017) Desmin loss and mitochondrial damage precede left ventricular systolic failure in volume overload heart failure. Am J Physiol Heart Circ Physiol 313(1):H32–Hh45
Ma LL, Ma X, Kong FJ et al (2018) Mammalian target of rapamycin inhibition attenuates myocardial ischaemia-reperfusion injury in hypertrophic heart. J Cell Mol Med 22(3):1708–1719
Schiattarella GG, Hill JA (2016) Therapeutic targeting of autophagy in cardiovascular disease. J Mol Cell Cardiol 95:86–93
Song HP, Chu ZG, Zhang DX et al (2018) PI3K-AKT pathway protects cardiomyocytes against hypoxia-induced apoptosis by MitoKATP-mediated mitochondrial translocation of pAKT. Cell Physiol Biochem 49(2):717–727
Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114(Pt 16):2903–2910
Zhang D, Contu R, Latronico MV et al (2010) MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest 120(8):2805–2816
Bulley SJ, Droubi A, Clarke JH et al (2016) In B cells, phosphatidylinositol 5-phosphate 4-kinase-alpha synthesizes PI(4,5)P2 to impact mTORC2 and Akt signaling. Proc Natl Acad Sci USA 113(38):10571–10576
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81670204); National Natural Science Foundation of China (Grant No. 81171839); Funding of Xiamen University (Grant No. 20720170106).
Author information
Authors and Affiliations
Contributions
Dongdong Sun, Lichao Hou, Erfei Zhang, and Xiaoying Zhao designed the experiments, analyzed and interpreted the data and drafted the manuscript. Li Zhang, Nan Li, Jingqi Yan, Ke Tu, Ruhu Yan, Jianqiang Hu and Mingming Zhang, were involved in the data acquisition. All authors revised the manuscript critically and approved the final version to be published. Dongdong Sun and Lichao Hou are responsible for the integrity of the work as a whole.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, E., Zhao, X., Zhang, L. et al. Minocycline promotes cardiomyocyte mitochondrial autophagy and cardiomyocyte autophagy to prevent sepsis-induced cardiac dysfunction by Akt/mTOR signaling. Apoptosis 24, 369–381 (2019). https://doi.org/10.1007/s10495-019-01521-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-019-01521-3