
Abstract
The brainstem has been a focus of sudden infant death syndrome (SIDS) research with amassing evidence of increased neuronal apoptosis. The present study extends the scope of brain regions examined and determines associations with known SIDS risk factors. Immunohistochemical expression of cell death markers, active caspase-3 and TUNEL, was studied in 37 defined brain regions in infants (aged 1–12 months) who died suddenly and unexpectedly (SUDI). A semi-quantitative mean score of marker expression was derived for each region and scores compared between three SUDI subgroups: explained SUDI (eSUDI; n = 7), SIDS I (n = 8) and SIDS II (n = 13). In eSUDI, active caspase-3 scores were highest in several nuclei of the rostral medulla, and lowest in the hypothalamus and cerebellar grey matter (GM). TUNEL was highest in regions of the hippocampus and basal ganglia, and lowest in the thalamus and cerebellar GM. TUNEL scores were higher in SIDS II compared to eSUDI in the amygdala (p = 0.03) and 5/9 nuclei in the rostral medulla (p = 0.04 − 0.01), and higher in SIDS II compared to SIDS I in the amygdala (p < 0.01), putamen (p = 0.01), lentiform nucleus (p = 0.03) and parietal (p = 0.03) and posterior frontal (p = 0.02) cortex. Active caspase-3 was greater in the hypoglossal nucleus (p = 0.03) of SIDS I compared to eSUDI infants. Co-sleeping, cigarette smoke exposure and the presence of an upper respiratory tract infection in SIDS infants was associated with differences in marker expression. This study affirms the sensitivity of the brainstem medulla to cell death in SIDS, and highlights the amygdala as a new region of interest.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sudden unexpected death in infancy (SUDI) encompasses all deaths of a sudden, unexpected nature in infants aged between 1 week and 1 year [1]. Following autopsy, SUDI deaths may be either explained (eSUDI), for example sudden death due to trauma, or remain unexplained, such as in the case of sudden infant death syndrome (SIDS) and those cases which are deemed undetermined/unascertained [2]. SIDS has been defined as “the sudden unexpected death of an infant < 1 year of age, with onset of the fatal episode apparently occurring during sleep, that remains unexplained after a thorough investigation, including performance of a complete autopsy and review of the circumstances of death and the clinical history” [3]. For the purposes of standardisation, the San Diego Criteria [3] may be used to sub-categorise SIDS into three groups: SIDS IA, cases wholly meeting the aforementioned Krous et al., definition with all necessary investigations performed but providing no insight into factors potentially contributing to death; SIDS IB, cases meeting most of the requirements of SIDS IA; however, the results of one or more of the post-mortem screening tests (toxicology, microbiology, radiology, vitreous chemistry and metabolic screening) are incomplete. SIDS II cases are those outside of the age of SIDS, or where potentially aggravating factors are present, including unsafe sleeping conditions, resolved developmental delays or acute illness, with these factors insufficient to result in death on their own.
In 2017, the Global Action and Prioritization of Sudden Infant Death Project identified “research on the role of abnormal or immature brain anatomy and physiology” to be an ongoing priority in SUDI research [4]. To date, the brainstem, which contains nuclei pertinent to the control of cardio-respiratory function, has been a focal point of SIDS neuropathology research, with multiple abnormalities being reported, predominantly in the medulla [reviewed: 5]. Notably, there is evidence of increased apoptosis in the brainstem of SIDS infants [6,7,8,9,10]. The present study, aims to align itself with the priority from the 2017 consensus, extending upon the literature to determine if there is any variation in expression of cell death markers in the wider central nervous system (CNS) of unexplained and explained SUDI infants.
Neuronal programmed cell death (PCD) is the active removal of selected neurons and is crucial for correct tissue development and homeostasis [11, 12]. PCD plays three key roles in the developing infant CNS: (i) restriction of progenitor cell numbers during neurogenesis; (ii) removal of defective neurons and; (iii) control of accurate connections and afferent/ efferent neuron ratios during synaptogenesis [11, 12]. PCD is intrinsically coupled with other developmental processes such as neuro- and synaptogenesis [11, 13]. The extent, timing and mechanisms of PCD have been reported to vary in different regions of the developing brain. This variation has been attributed to the differential progression of concurrent developmental processes (i.e. neurogenesis and synaptogenesis) as well as several other factors, including the balance of pro-survival and pro-death factors in the local microenvironment, neuron type, metabolic demands and vascular supply [14,15,16]. While processes critical to development are occurring, the immature brain is more sensitive to insult [15].
Apoptosis is the predominant form of PCD active in the CNS during development and is mediated by a family of intracellular cysteine proteases (caspases) [17]. Although initially thought to be specific for apoptosis, terminal deoxynucleotidyl transferase (Tdt)-mediated dUTP-biotin nick end-labelling (TUNEL) identifies DNA fragmentation in multiple cell death pathways including necrosis and apoptosis [18]. Active caspase-3 is frequently used as a specific marker of apoptosis. In the SIDS brain, an increase in TUNEL has been reported in the hippocampus [10] and in the arcuate, cuneate and gracile nuclei of the medulla [8, 10, 19]. Increased active caspase-3 expression has also been identified in the medulla in two independent SIDS cohorts [6,7,8], however, neither TUNEL nor active caspase-3 expression were increased in the hypothalamus [20], suggesting that increased cell death in the SIDS brain is not global. To better understand the cell death paradigm in the infant brain, the present study focused on extending the brain regions examined, with analysis comparing three sub-groups of SUDI (eSUDI, SIDS I and SIDS II). The findings are intended to help direct future neuropathology studies by identifying any further potential regions of interest in the SUDI brain, and to help address the priorities of the international consensus on SUDI research.
Materials and methods
Data and tissue collection
All information regarding infant characteristics and the brain tissue used in this study were collected by the Department of Forensic Medicine, NSW between 2008 and 2012. The data for this study were extracted from the autopsy report including the neuropathology report and associated clinical information collected by the Department of Forensic Medicine. Additional information pertaining to tissue handling, fixation method and duration was also obtained in order to identify any variations which might have implications on histological staining. This study had ethical approval from the Sydney Local Health District NSW (RPAH Zone) and University of Sydney Ethics review committees, and from the NSW State Coroner. In accordance with ethical guidelines, all identifying data were removed and case numbers were used to identify tissue samples and associated infant characteristics.
7 µm sections of 20% formalin fixed paraffin embedded tissue were mounted on 2% 3-aminopronopyltriethoxysilane treated slides. Wherever possible, a minimum of two serial sections were collected for staining with the two markers. Due to limited tissue availability, duplicate staining was performed for only 20% of sections examined. Thirty-seven macro- or microscopically defined regions were defined for examination. Briefly, this included: the frontal, parietal, occipital, temporal and insular cortex, basal ganglia structures (the striatum and lentiform nucleus) and their components (head of the caudate nucleus, putamen, globus pallidus, claustrum), amygdala, anterior thalamic nuclear group, the lateral geniculate nucleus, hypothalamus, hippocampus, the dentate nucleus, white matter and three grey matter layers of the cerebellar hemisphere and nine nuclei of the rostral medulla (arcuate, cuneate, dorsal motor nucleus of the vagus, inferior olivary, lateral reticular formation, nucleus of the solitary tract, nucleus of the spinal trigeminal tract, the vestibular (medial and spinal combined) and hypoglossal nucleus).
Immunostaining
Peroxidase immunohistochemistry for active caspase-3 and TUNEL was performed manually as previously described [6]. All steps were performed at room temperature unless otherwise stated. In brief, sections were deparaffinised, rehydrated and subjected to microwave antigen retrieval in TRIS–EDTA buffer (1 mM trisodium citrate, 1 mM EDTA, 2 mM TRIS; pH 9.0) and cooled. Endogenous peroxidase activity was quenched in a 3% hydrogen peroxide, methanol and phosphate buffered saline (PBS) solution.
Sections for active caspase-3 staining were blocked for 1 h in 10% normal horse serum (NHS) and incubated overnight in affinity purified rabbit anti-active caspase-3 antibody (1:200; Cat no: 559565, BD Pharmigen). After washing with PBS, sections were incubated for 1 h in secondary antibody (anti-rabbit IgG; 1:250; BA-1000, Vector Laboratories). This was followed by further washing before incubation in a peroxidase avidin–biotin complex (ABC) (PK-4000, Vectastain ABC Kit, Vector Laboratories) for 1 h.
TUNEL staining was performed using a commercially available kit (Millipore, ApopTag Peroxidase in Situ kit, #S7100), with slight modification to the manufacturer’s instructions. Sections were covered briefly in equilibration buffer prior to incubation in a humidified chamber for 1 h at 37 °C with the Tdt-enzyme (diluted 1:2:1-Tdt: reaction buffer: dH2O). The labelling reaction was stopped by 10-min immersion in stop-wash buffer. Sections were incubated in anti-digoxygenin-peroxidase for 40 min and washed in PBS.
Following final PBS washes, both active caspase-3 and TUNEL stained sections were incubated with chromogen (3,3′-diaminobenzidine [DAB]) and counterstained with Harris’ haematoxylin. Sections were dehydrated through a graded ethanol series and cleared in xylene before mounting.
Negative controls for IHC were incubated with 1% NHS in place of primary antibody (active caspase-3), and for TUNEL, negative control sections were incubated in the absence of Tdt (2:2-reaction buffer: dH2O).
For the hippocampus, lateral geniculate nucleus (LGN) and rostral medulla, active caspase-3 and TUNEL staining were double stained. TUNEL colour development with DAB preceded blocking of sections in 10% NHS and active caspase-3 staining as above. However, following incubation with alkaline phosphatase ABC (AK-5000, Vectastain ABC Kit, Vector Laboratories), sections were washed in 0.05 M Tris-buffered saline (pH 7.5) and colour developed in the dark for 45 min in nitro blue tetrazolium chloride (NBT) (K0598, Dako North America Inc.) prior to cover-slipping using VectMount (H-5000, Vector Laboratories). For these sections, nuclear TUNEL and cytoplasmic active caspase-3, were scored independently. To ensure comparability between single and double labelled sections, 20% of the co-stained sections also had separate staining for active caspase-3 and TUNEL.
Semi-quantitative scoring of immunostaining
Sections were examined using the 20× objective lens of a Leica Upright DM6000 Light Microscope (Leica Microsystems Ltd. Heerbrugg, Switzerland). Blind to diagnosis, a single scorer, counted the number of neurons positive for active caspase-3 or TUNEL in each of the 37 defined areas and expressed these counts as a percentage of the total number of neurons in each area. A semi-quantitative score from 1 to 5 was derived as follows: ‘0’ = 0%; ‘1’ = 1–20%; ‘2’ = 21–50%; ‘3’ = 51–70% and ‘4’ > 70% of neurons with positive marker expression in a given region. This semi-quantitative method has been used previously by our group [7, 10].
Statistical analysis
All data were exported to SPSS for Windows (version 21; SPSS (IBM) Inc., Illinois, USA) for statistical analysis. ANOVA with post-hoc least significant difference (LSD) correction was used to compare continuous infant characteristic data and the mean scores for immunostaining between diagnostic groups; these values are presented as the mean ± standard error of the mean (SEM). Independent samples t-tests were used to compare mean scores of marker expression in the presence and absence of risk factors. Chi square testing (Fischer’s exact test) was used to compare nominal infant data characteristics; these values are expressed as a percentage. A total mean score (taken across all 37 regions) was also calculated for active caspase-3 and TUNEL for each case and used for statistical comparison between eSUDI, SIDS I and SIDS II infants. For all tests, significance was taken at p < 0.05.
Results
Data characteristics
The 28 cases included in this study are a subset of the previously characterised 2008–2012 SIDS cohort [6]. All of the eSUDI (n = 7) and SIDS I (n = 8) cases of the dataset are included herein, and only 13/49 SIDS II cases; selection was on the basis of sex and age matching of SIDS II cases to eSUDI infants and tissue section availability. For the purposes of analysis, SIDS IA (n = 4) and SIDS IB (n = 4) cases were pooled together as SIDS I (n = 8). Diagnoses of the eSUDI infants were: acute pneumonia (n = 1), thrombotic occlusion of Blalock Taussig Shunt (n = 1), complication of congenital heart disease (n = 2), myocarditis (n = 2), and meningoencephalitis of uncertain aetiology (n = 1).
The three diagnostic groups were matched for clinical and autopsy features and the prevalence of SIDS risk factors. Consistent with diagnostic criteria, the presence of bed-share/co-sleeping was greater in SIDS II compared to SIDS I infants (69% vs 0% p < 0.01) (Table 1).
Immunostaining and analysis
Marker expression in the eSUDI infant brain
Active caspase-3 (Table 2)
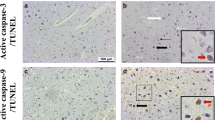
Positive active caspase-3 staining was restricted to the cytoplasm of neurons (Fig. 1a–d). The mean score of active caspase-3 ranged from zero in the head of the caudate nucleus, the hypothalamus and the cerebellar cortex (internal granular layer, Purkinje cells and molecular layer) to 2.1 ± 0.1 in the arcuate, cuneate and inferior olivary nuclei in the rostral medulla and was < 1 in 30 of the 37 regions examined. Active caspase 3 staining was identified in the globus pallidus and the arcuate, cuneate and inferior olivary nuclei in the rostral medulla of all eSUDI infants (Table 2).

Micrographs demonstrating active caspase-3 and TUNEL expression in the infant brain. Active caspase-3 (a–d) and TUNEL (e–h) expression is shown in the dentate nucleus of the cerebellum (a, e), putamen (b, f), amygdala (c, g) and CA1 region of the hippocampus (d, h). Solid black arrows indicate positive neurons and thin white arrows indicate negative neurons. Scale bar represents 50 µm for the dentate nucleus (a, e) and 20 µm for all other regions (b–d, f–h). CA cornu ammonis, TUNEL terminal deoxynucleotidyl transferase (Tdt)-mediated dUTP-biotin nick end-labelling
TUNEL (Table 3 )
Positive TUNEL staining was restricted to the nucleus of neurons with many showing the typical features of cell death, including shrinkage and pyknosis (Fig. 1e–h). The mean score of TUNEL ranged from zero in the Purkinje cells of the cerebellum and anterior nuclei of the thalamus to 2.0 ± 0.3 in the entorhinal cortex of the hippocampus, with the mean score < 1 in 23 of the 37 brain regions examined. TUNEL staining was identified in the lentiform nucleus, dentate gyrus, entorhinal cortex, subiculum and CA4 region of the hippocampus in all eSUDI infants (Table 3).
TUNEL compared to active Caspase-3
In 24 of the 37 brain regions, the mean score of TUNEL was greater than the mean score of active caspase-3. The total mean scores (taken across all 37 brain regions) of active caspase-3 (0.6 ± 0.1) and TUNEL (0.8 ± 0.1) were not statistically different (Fig. 2).

Diagnostic group comparisons of mean total score of cell death marker expression across all thirty-seven brain regions examined. Scoring schema: 0 = 0%; 1 = 1–20%; 2 = 21–50%, 3 = 51–70%; 4 > 70% neurons with positive staining in the given region. Blue line indicates the mean total score of marker expression, regardless of diagnosis. *Significance taken at p < 0.05 following one-way ANOVA with post-hoc LSD correction. eSUDI explained sudden unexpected death in infancy, SIDS sudden infant death syndrome, TUNEL terminal deoxynucleotidyl transferase (Tdt)-mediated dUTP-biotin nick end-labelling
Differences between diagnostic groups (eSUDI vs SIDS I vs SIDS II)
Active caspase-3 (Table 2) and TUNEL (Table 3)
Comparing diagnostic groups, a difference in the mean score of active caspase-3 was seen in 1 of the 37 brain regions, with SIDS I infants having a greater mean score of active caspase-3 compared to eSUDI infants in the hypoglossal nucleus (XII) (p = 0.03). The mean scores of active caspase-3 in the CNS of SIDS II infants were not different compared to eSUDI and SIDS I infants.
Compared to eSUDI infants, SIDS II infants had a significantly greater mean score of TUNEL in the amygdala (p = 0.03) and the arcuate (p < 0.01), cuneate (p < 0.01), inferior olivary (p = 0.01), lateral reticular formation (p = 0.04) and hypoglossal (p = 0.02) nuclei of the rostral medulla. Compared to SIDS I, SIDS II infants had greater mean scores of TUNEL in the: amygdala (p < 0.01), lentiform nucleus (p = 0.03), putamen (p = 0.01), and the posterior frontal (p = 0.02) and parietal (p = 0.03) cortex. The mean score of TUNEL was greater in the CA1 region of the hippocampus in SIDS I compared to SIDS II infants (p < 0.01).
Mean total score apoptotic marker expression (Fig. 2)
The mean total score for active caspase-3, across all 37 brain regions, was similar in all three diagnostic groups. The mean total score of TUNEL was significantly greater in SIDS II compared to SIDS I infants (p < 0.01).
Risk factor analysis
Risks independent of diagnostic group (Table 4)
The impact of cigarette smoke exposure and the presence of an URTI on cell death marker expression was analysed across all infants in the dataset, regardless of diagnosis (where data was available).
Infants with cigarette smoke exposure (n = 8) had greater mean scores of active caspase-3 than those without (n = 15) in the occipital cortex (p = 0.03), hypoglossal nucleus (p = 0.04), the CA1 (p < 0.01) and CA4 (p = 0.01) regions of the hippocampus. In the dentate nucleus, the mean score of TUNEL was greater in infants with a positive history of cigarette smoke exposure compared to those with a negative history (p = 0.03).
Compared to infants without an URTI (n = 19), infants with an URTI (n = 9) in the 2 weeks prior to death had a greater mean score of active caspase-3 in the NSTT (p < 0.01). Infants without an URTI had greater mean scores of active caspase-3, compared to infants with an URTI in the anterior superior frontal (p = 0.03) and insular (p = 0.02) cortex. Compared to infants without an URTI, infants with an URTI had greater mean scores of TUNEL in the amygdala (p = 0.03), dentate gyrus (p = 0.02), parietal cortex (p = 0.01) and the arcuate (p = 0.04), cuneate (p < 0.01), lateral reticular formation (p = 0.02), nucleus of the spinal trigeminal tract (p < 0.01), nucleus of the solitary tract (p = 0.04), and vestibular (p < 0.01) nuclei of the rostral medulla.
Sleep related risk factors—SIDS infants only (Table 5)
Sleep related risk factors (being found in the prone sleeping position and bed-sharing/co-sleeping), were analysed in SIDS infants (SIDS I and SIDS II combined). Bed share/co-sleeping was associated with a greater mean score of active caspase-3 in the lentiform nucleus (p = 0.04) and the cuneate nucleus (p = 0.01). No differences in the mean score of TUNEL were observed between bed sharers/co-sleepers (n = 9) and non-bed sharers/co-sleepers (n = 4). Compared to infants found in other sleep positions (n = 11), infants found in the prone sleeping position had greater mean scores of active caspase-3 in the striatum (p = 0.03), subiculum (p = 0.03) and dentate nucleus (p = 0.02). No differences in the mean score of TUNEL were observed between infants found in the prone sleeping position and infants found in other positions.
Discussion
The main findings of this study include: (1) In the developing infant brain, PCD is highest in the medulla and hippocampus, but minimal in the cerebellum. (2) SIDS II had higher expression of TUNEL compared to eSUDI infants in the five nuclei of the rostral medulla and in the amygdala, and compared to SIDS I infants in the lentiform nucleus and the parietal and posterior frontal cortex. (3) A single difference was observed for active caspase-3 with greater expression in the hypoglossal nucleus in SIDS I compared to eSUDI infants. (4) The risk factors of a presence of an URTI and cigarette smoke exposure, were associated with greater levels of cell death amongst a greater number of brain regions when compared to the sleep related risks of prone sleeping or bed-sharing.
Cell death marker expression in the eSUDI infant brain
The first component of the present study was the exploratory examination of the distribution of cell death markers in the eSUDI brain. To the best of our knowledge, the pattern of physiological PCD in the human brain between the end of the first month and the end of the first year of life, has not previously been profiled in as many regions. In the human postnatal brain, PCD has been shown to occur in regions with active neurogenesis, such as the external granular cell layer (EGL) of the cerebellum and the dentate gyrus of the hippocampus, as well as in post-mitotic neurons at sites of synaptogenesis, spread throughout the cerebral cortex and cerebellum [11, 13, 21, 22]. In the present study, PCD was not restricted to certain regions of the brain, rather varied across the CNS, with the cerebellum, hypothalamus and structures of the basal ganglia demonstrating some of the lowest scores of cell death marker expression and sub-regions of the medulla and hippocampus demonstrating some of the highest scores. Regional heterogeneity likely reflects the fine balance between pro- and anti- cell survival factors in the local microenvironment, neuron type and stage of maturation [14,15,16].
The dentate gyrus of the hippocampus, a site of postnatal neurogenesis, demonstrated one of the greatest mean scores of TUNEL of all brain regions. This finding is consistent with Sparks et al., who reported Alz-50 (a marker associated with neurons dying through degenerative processes) expression in the non-SIDS para-hippocampal gyrus [23]. The TUNEL study by Waters et al. did not examine the dentate gyrus [10].
Of all the brain regions examined, the cerebellar cortex and dentate nucleus have the lowest expression of both active caspase-3 and TUNEL. The EGL of the cerebellum is a key site of postnatal neurogenesis and is present from the 9–11th gestational week to the 11th postnatal month in human infants [24], decreasing in thickness during the second half of the first year of life [24]. Given the age-related variation in EGL presence and/or thickness, which is quite marked over the first year of life, this region was excluded from examination in the present study. PCD in the human cerebellum is reported to be greatest in the EGL, peaking at postnatal day 3 [25]. In the same study, compared to the EGL, lower levels of cell death were observed in the internal granular layer, molecular layer and cerebellar white matter, with each peaking in the first postnatal month and falling to near zero by the fourth postnatal month in humans [25]. Thus, the low levels of cell death markers seen in the cerebellum of our eSUDI infants (mean age 4.1 months) are consistent with the literature.
Nuclei of the rostral medulla, including the arcuate, cuneate and inferior olivary nuclei, demonstrated the greatest mean scores of active caspase-3 expression of all brain regions. The heterogeneity in active caspase-3 and TUNEL expression amongst the nuclei of the rostral medulla was expected given previous findings of different levels of neuronal death amongst medullary nuclei during development [26, 27]. Similar to our findings, very low TUNEL expression had been demonstrated in the hypoglossal nucleus of infants in their first year of life by Porzionato et al. [26]. We identified the greatest TUNEL score in the arcuate nucleus, however this region was not examined by Porzionato et al. [26].
SIDS compared to eSUDI
We identified differences in either activated caspase-3 or TUNEL scores in 11 of the 37 brain regions examined. Of these, five differences were in the rostral medulla and confirm previously reported results [6,7,8,9,10], despite the current study being on a smaller sample size and including a proportion of the SIDS II population studied by Anbrose et al., 2018 [6]. The hypoglossal nucleus was the only region to demonstrate greater active caspase-3 in the SIDS population of this study, and also had greater TUNEL; this is consistent with two of our previous studies [7, 8]. Present findings add to the data reporting abnormalities of the SIDS hypoglossal nucleus including increased number of synapses [28] and morphological changes [29]. In the present study, greater mean scores of active caspase-3 in the hypoglossal nucleus were associated with cigarette smoke exposure, and cigarette exposure was similarly associated with the morphological changes reported in the hypoglossal nucleus of eSUDI infants by Lavezzi et al. [29]. Functionally, the hypoglossal nucleus is involved in maintaining airway patency via motor control of the tongue muscles [30]. Animal studies have shown causal association between nicotine exposure and perturbed hypoglossal function [31,32,33]. Taken together, cigarette smoke exposure may be a factor contributing to the hypoglossal abnormalities amassing in the literature; consequently, putting the infant at risk of death due to diminished airway patency.
The amygdala was the only region demonstrating heightened TUNEL in SIDS II infants compared to both eSUDI and SIDS I infants. The amygdala is a telencephalic component of the limbic system situated in the dorsomedial temporal lobe [34]. Through a myriad of afferent and efferent projections, the amygdala is implicated in integrative responses to affective behaviour and environmental cues, and via connections to the hippocampus is involved in episodic memory processing [34, 35]. The significance in secondary cell death changes to this area, in the context of SIDS are yet to be elucidated, however abnormal changes in the amygdala have been implicated in a number of neurodevelopmental disorders [reviewed: 35]. Recently, the amygdala has been identified as an integral structure in the central homeostatic network of the human brain having close connections with the brainstem [36], with specific links to the hypoglossal nucleus and dorsal motor nucleus of the vagus in the medulla [37]. A limitation of the present study is examination of the amygdala complex as a single entity and not sub-analysis of its individual nuclei. Thus, future studies should consider examining individual nuclei of the amygdala, in turn exploring the relevant functional implications in the context of SIDS.
With adherence to the San Diego criteria, SIDS I cases are seen to be “classic SIDS” and to represent a population with fewer SIDS risk factors as compared to SIDS II infants. The CA1 region of the hippocampus is the only region to demonstrate greater TUNEL scores in SIDS I compared to SIDS II infants. Given, there were no changes in TUNEL expression reported in the presence of the SIDS risk factors analysed, this difference likely reflects a change more closely related to the pathogenesis of SIDS as opposed to confounding risk factor presence.
Hypoxic-ischemia (HI) is a cell death stimulus that has been extensively discussed in the context of SIDS and is commonly associated with the caspase-9 and caspase-3 mediated intrinsic apoptotic pathway, however neuronal death following HI may also be attributed to necrotic processes [17, 38,39,40]. The nature of the cell death stimulus and upstream signals, along with availability of downstream substrates dictate the cell death pathways implicated [41, 42] and potentially contributes to the observed variation in cell death marker expression amongst brain regions. A recent study by our laboratory found no change in active caspase-9; questioning its involvement in the SIDS medulla [6]. In the present study, differences between diagnostic groups were more evident with TUNEL, than with active caspase-3, staining. DNA fragmentation, identified with TUNEL is a common end-point point of multiple cell death pathways [18]. The greater TUNEL expression in SIDS II infants, without an accompanying increase in active caspase-3 suggests non-apoptotic cell death mechanisms are potentially implicated and highlights the complexity of cell death processes in SIDS and the need to better understand and identify both the cell death stimulus and the pathway(s) involved. The overall lack of significant difference in TUNEL expression between eSUDI and SIDS I, in all thirty-seven brain regions, suggests an inherent similarity in the amount and/or type of exposure to stimuli promoting DNA fragmentation and cell death in both groups. A limitation of this study, as with all SIDS research, that might contribute to this observation is the limited and heterogeneous composition of the eSUDI control group.
Risk factor association with cell death markers
The impact of identifiable risk factors on cell death marker expression were evaluated. The greater TUNEL scores seen in SIDS II infants compared to the eSUDI and/or SIDS I infants may reflect the presence of confounding risk factor(s) in the SIDS II group. Of the four SIDS risk factors analysed, only bed share/co-sleeping differentiates SIDS II from SIDS I cases and is reflected in the statistical difference observed between risk profiles for SIDS I and SIDS II infants [3]. In the present study, the mean score of active caspase-3 was greater in the lentiform nucleus and cuneate nucleus of bed sharers/co-sleepers, and greater in the striatum, subiculum and dentate nucleus of infants found in the prone sleeping position. When stratifying for the presence and absence of these sleep related SIDS risk factors, no differences were seen in TUNEL scores, this contrasts with our previous finding of increased TUNEL in the cuneate and gracile nuclei of alone sleepers [8]. Caspase-3 activation can be detected within an hour of a HI insult, reaching peak levels after 18–24 h [43, 44] and is followed by DNA fragmentation (TUNEL expression) peaking 24 h after insult [45, 46]. Thus, the heightened active caspase-3, in the absence of changes in TUNEL, suggests a recent hypoxic-ischemic insult, attributable to sleep related risk factors [43,44,45,46].
Cigarette smoke exposure is a leading modifiable SIDS risk factor [47] but also predisposes infants to other illnesses including respiratory infections [48]. In the present study, regardless of diagnosis, exposure to cigarette smoke was associated with greater active caspase-3 in the occipital cortex, hypoglossal nucleus, CA1 and CA4 region, and greater TUNEL in the dentate nucleus. Previous studies have also identified increased TUNEL expression in the AN and dorsal motor nucleus of the vagus in infants exposed to cigarette smoke exposure [8], although this was not observed in the present study. The difficulties in reproducing (and interpreting) these findings are not surprising given the variabilities possible in type (pre- vs post-natal, active and passive) and duration of exposure to cigarette smoke that may exist and could contribute to differences in the expression of apoptotic markers and potential mechanisms underpinning neuronal death. Regardless, the combined findings in the literature provide evidence that cigarette smoke exposure does, in itself, lead to upregulation of cell death pathways and this is not restricted to the brainstem medulla. This is further supported by work in a mouse pre-into post-natal cigarette smoke exposure model, where increased active caspase-3 and TUNEL were reported in several nuclei of the medulla [49] and hippocampus [50].
In the present study, a history of an URTI was associated with greater TUNEL expression in the amygdala, dentate gyrus, parietal cortex and six of the nine nuclei of the rostral medulla; this included the nucleus of the spinal trigeminal tract, a nucleus previously reported to demonstrate an association between URTI and increased cell death marker expression [6]. An URTI was reported in 46% of SIDS II infants, thus the potential contribution of this risk factor to heightened TUNEL levels, as seen in SIDS II compared to eSUDI and SIDS I, cannot be discounted. The observed increase in neuronal cell death following an URTI is likely a secondary effect of the inflammatory response mounted towards the systemic immune challenge. Following peripheral infection there is increased systemic circulation of monocytes, cytokines, kinins and other mediators of inflammation, and these have the potential to reach the brain [51]. In neurodegenerative disorders, it has been suggested that inflammatory mediators following systemic challenge, accelerate or exacerbate underlying neurodegenerative processes [51, 52]. Although further investigation into the mechanisms which underpin these observations are necessary, it is suggested that events at the immune-system-brain interface result in CNS localised inflammatory responses, such as microglial activation, which exacerbate local pathological events [51, 52]. Microglia have been shown to interact with neurons, selectively targeting extra-numerary, distressed and/or dying neurons [53]. Aberrant microglia activation and subsequent pro-inflammatory responses have been reported to promote collateral tissue destruction [54]. While involvement of circum-ventricular organs might have been expected, given they are one proposed site of the immune-system-brain interface [52], this was not the case, making it difficult to explain why in the present study, heightened cell death was in particular brain regions following an URTI. Overall, these findings suggest neuronal cell death processes occurring in SIDS infants are further exacerbated by the presence of systemic inflammatory mediators following an URTI, which upon reaching the CNS, they promote local inflammatory responses that facilitate and/or accelerate cell death pathways.
Conclusion
The present study is the first to profile apoptosis amongst 37 defined regions of the human infant (1–12 months of age) brain, and confirms that the expression of PCD markers (active caspase-3 and TUNEL) is non-homogeneous across these regions. When comparing infants who died suddenly, regional differences were seen amongst those with an explained cause of death, SIDS I and SIDS II, with five of the eleven observed differences being localised to the rostral medulla of the brainstem. Furthermore, known SIDS risk factors such as co-sleeping, cigarette smoke exposure and a recent URTI were associated with differences in the expression of the PCD markers studied.
Abbreviations
- AN:
-
Arcuate nucleus
- CA:
-
Cornu ammonis
- CNS:
-
Central nervous system
- Cun:
-
Cuneate nucleus
- DMNV:
-
Dorsal motor nucleus of the vagus
- eSUDI:
-
Explained sudden unexpected death in infancy
- ION:
-
Inferior olivary nucleus
- LRt:
-
Lateral reticular formation
- NSTT:
-
Nucleus of the spinal trigeminal tract
- PCD:
-
Programmed cell death
- SIDS:
-
Sudden infant death syndrome
- SUDI:
-
Sudden unexpected death in infancy
- TUNEL:
-
Terminal deoxynucleotidyl transferase (Tdt)-mediated dUTP-biotin nick end-labelling
- URTI:
-
Upper respiratory tract infection
- Vest:
-
Medial and spinal vestibular nuclei combined
- XII:
-
Hypoglossal nucleus
References
Fleming P, Blair P, Bacon C, Berry J (2000) Sudden unexpected deaths in infancy: the CESDI SUDI studies 1993–1996. The Stationery Office, London, pp 1–5
Blair PS, Byard RW, Fleming PJ (2012) Sudden unexpected death in infancy (SUDI): suggested classification and applications to facilitate research activity. Forensic Sci Med Pathol 8:312–315
Krous HF, Beckwith JB, Byard RW et al (2004) Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics 114:234–238
Hauck FR, McEntire BL, Raven LK et al (2017) Research priorities in sudden unexpected infant death: an international consensus. Pediatr 140:e20163514
Machaalani R, Waters KA (2014) Neurochemical abnormalities in the brainstem of the Sudden Infant Death Syndrome (SIDS). Paediatr Respir Rev 15:293–300
Ambrose N, Waters KA, Rodriguez ML, Bailey K, Machaalani R (2018) Neuronal apoptosis in the brainstem medulla of sudden unexpected death in infancy (SUDI), and the importance of standardized SUDI classification. Forensic Sci Med Pathol 14:42–56
Machaalani R, Rodriguez M, Waters K (2007) Active caspase-3 in the sudden infant death syndrome (SIDS) brainstem. Acta Neuropathol 113:577–584
Machaalani R, Waters KA (2008) Neuronal cell death in the Sudden Infant Death Syndrome brainstem and associations with risk factors. Brain 131:218–228
Sparks LD, Hunsaker JC (2002) Neuropathology of sudden infant death (syndrome): literature review and evidence of a probable apoptotic degenerative cause. Childs Nerv Syst 18:568–592
Waters KA, Meehan B, Huang J, Gravel RA, Michaud J, Côté A (1999) Neuronal apoptosis in sudden infant death syndrome. Pediatr Res 45:166–172
Buss RR, Sun W, Oppenheim RW (2006) Adaptive roles of programmed cell death during nervous system development. Annu Rev Neurosci 29:1–35
Dekkers MP, Nikoletopoulou V, Barde Y-A (2013) Death of developing neurons: New insights and implications for connectivity. J Cell Biol 203:385–393
Oppenheim RW (1991) Cell death during development of the nervous system. Annu Rev Neurosci 14:453–501
Pfisterer U, Khodosevich K (2017) Neuronal survival in the brain: neuron type-specific mechanisms. Cell Death Dis 8:e2643
Rice D, Barone S Jr (2000) Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect 108:511
Lossi L, Merighi A (2003) In vivo cellular and molecular mechanisms of neuronal apoptosis in the mammalian CNS. Prog Neurobiol 69:287–312
Elmore S (2007) Apoptosis: a review of programmed cell death. J Toxicol Pathol 35:495–516
Häcker G (2000) The morphology of apoptosis. Cell Tissue Res 301:5–17
Sparks LD, Hunsaker JC (2002) Neuropathology of sudden infant death (syndrome): literature review and evidence of a probable apoptotic degenerative cause. Child’s Nervous System 18:568–592
Hunt NJ, Waters KA, Machaalani R (2016) Promotion of the unfolding protein response in orexin/dynorphin neurons in sudden infant death syndrome (SIDS): elevated pPERK and ATF4 expression. Mol Neurobiol 54:1–15
Blaschke AJ, Weiner JA, Chun J (1998) Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J Comp Neurol 396:39–50
Kim WR, Sun W (2011) Programmed cell death during postnatal development of the rodent nervous system. Dev Growth Differ 53:225–235
Sparks DL, Davis DG, Mellert Bigelow T et al (1996) Increased ALZ-50 immunoreactivity in sudden infant death syndrome. J Child Neurol 11:101–107
Ábrahám H, Tornóczky T, Kosztolányi G, Seress L (2001) Cell formation in the cortical layers of the developing human cerebellum. Int J Dev Neurosci 19:53–62
Lossi L, Zagzag D, Greco MA, Merighi A (1998) Apoptosis of undifferentiated progenitors and granule cell precursors in the postnatal human cerebellar cortex correlates with expression of BCL-2, ICE, and CPP32 proteins. J Comp Neurol 399:359–372
Porzionato A, Macchi V, Guidolin D, Sarasin G, Parenti A, Caro RD (2008) Anatomic distribution of apoptosis in medulla oblongata of infants and adults. J Anatomy 212:106–113
Stecco C, Porzionato A, Macchi V et al (2004) Detection of apoptosis in human brainstem by TUNEL assay. Italian J Anat Embryol (Archivio italiano di anatomia ed embriologia) 110:255–260
O’kusky JR, Norman MG (1995) Sudden infant death syndrome: increased number of synapses in the hypoglossal nucleus. J Neuropathol Exp Neurol 54:627–634
Lavezzi AM, Corna M, Mingrone R, Matturri L (2010) Study of the human hypoglossal nucleus: normal development and morpho-functional alterations in sudden unexplained late fetal and infant death. Brain Dev 32:275–284
O’Kusky JR, Norman MG (1992) Sudden infant death syndrome: postnatal changes in the numerical density and total number of neurons in the hypoglossal nucleus. J Neuropathol Exp Neurol 51:577–584
Pilarski JQ, Wakefield HE, Fuglevand AJ, Levine RB, Fregosi RF (2010) Developmental nicotine exposure alters neurotransmission and excitability in hypoglossal motoneurons. J Neurophysiol 105:423–433
Powell GL, Levine RB, Frazier AM, Fregosi RF (2014) Influence of developmental nicotine exposure on spike-timing precision and reliability in hypoglossal motoneurons. J Neurophysiol 113:1862–1872
Jaiswal SJ, Buls Wollman L, Harrison CM, Pilarski JQ, Fregosi RF (2016) Developmental nicotine exposure enhances inhibitory synaptic transmission in motor neurons and interneurons critical for normal breathing. Dev Neurobiol 76:337–354
Nieuwenhuys R, Voogd J, Van Huijzen C (1988) The human central nervous system: a synopsis and atlas. Springer Science & Business Media, Berlin
Schumann CM, Bauman MD, Amaral DG (2011) Abnormal structure or function of the amygdala is a common component of neurodevelopmental disorders. Neuropsychologia 49:745–759
Edlow BL, McNab JA, Witzel T, Kinney HC (2016) The structural connectome of the human central homeostatic network. Brain Connect 6:187–200
Van Daele D, Fazan V, Agassandian K, Cassell M (2011) Amygdala connections with jaw, tongue and laryngo-pharyngeal premotor neurons. Neuroscience 177:93–113
Machaalani R, Waters KA (2003) Increased neuronal cell death after intermittent hypercapnic hypoxia in the developing piglet brainstem. Brain Res 985:127–134
Yue X, Mehmet H, Penrice J et al (1997) Apoptosis and necrosis in the newborn piglet brain following transient cerebral hypoxia–ischaemia. Neuropathol Appl Neurobiol 23:16–25
Feng Y, Fratkin JD, LeBlanc MH (2003) Inhibiting caspase-9 after injury reduces hypoxic ischemic neuronal injury in the cortex in the newborn rat. Neurosci Lett 344:201–204
Daugas E, Nochy D, Ravagnan L et al (2000) Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett 476:118–123
Susin SA, Daugas E, Ravagnan L et al (2000) Two distinct pathways leading to nuclear apoptosis. J Exp Med 192:571–580
Wang X, Zhu C, Wang X et al (2004) X-linked inhibitor of apoptosis (XIAP) protein protects against caspase activation and tissue loss after neonatal hypoxia–ischemia. Neurobiol Dis 16:179–189
Snider BJ, Gottron FJ, Choi DW (1999) Apoptosis and necrosis in cerebrovascular disease. Ann N Y Acad Sci 893:243–253
Cheng Y, Deshmukh M, D’Costa A et al (1998) Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Investig 101:1992
Zhu C, Wang X, Hagberg H, Blomgren K (2000) Correlation between Caspase-3 activation and three different markers of DNA damage in neonatal cerebral hypoxia-ischemia. J Neurochem 75:819–829
Mitchell EA, Freemantle J, Young J, Byard RW (2012) Scientific consensus forum to review the evidence underpinning the recommendations of the Australian SIDS and Kids Safe Sleeping Health Promotion Programme–October 2010. J Paediatr Child Health 48:626–633
DiFranza JR, Aligne CA, Weitzman M (2004) Prenatal and postnatal environmental tobacco smoke exposure and children’s health. Pediatrics 113:1007–1015
Vivekanandarajah A, Chan YL, Chen H, Machaalani R (2016) Prenatal cigarette smoke exposure effects on apoptotic and nicotinic acetylcholine receptor expression in the infant mouse brainstem. Neurotoxicology 53:53–63
Chan YL, Saad S, Machaalani R et al (2017) Maternal cigarette smoke exposure worsens neurological outcomes in adolescent offspring with hypoxic-ischemic injury. Front Mol Neurosci 10:306
Perry VH, Newman TA, Cunningham C (2003) The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci 4:103
Perry VH, Cunningham C, Holmes C (2007) Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 7:161
Tremblay M-È, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A (2011) The role of microglia in the healthy brain. J Neurosci 31:16064–16069
Rock KL, Latz E, Ontiveros F, Kono H (2010) The sterile inflammatory response. Ann Rev Immunol 28:321–342
Acknowledgements
The tissue used in this study was provided by the NSW Forensic and Analytical Science Service. The authors acknowledge the contribution of Arunnjah Vivekanandarajah in the staining of hippocampal sections. The authors acknowledge the facilities, and scientific and technical assistance of the Australian Microscopy and Microanalysis Research Faculty at the Australian Centre of Microscopy and Micro Analysis, University of Sydney.
Funding
This study was funded by philanthropy (The Miranda Belshaw Foundation, Australia).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Ethical approval
Ethical approval was from the NSW health RPAH zone (X13-0038 and HREC/13/RPAH/54) and University of Sydney Ethic committees.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ambrose, N., Rodriguez, M., Waters, K.A. et al. Cell death in the human infant central nervous system and in sudden infant death syndrome (SIDS). Apoptosis 24, 46–61 (2019). https://doi.org/10.1007/s10495-018-1509-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-018-1509-0