
Abstract
Lung cancer has a relatively poor prognosis with a low survival rate and drugs that target other cell death mechanism like autophagy may help improving current therapeutic strategy. This study investigated the anti-proliferative effect of Licarin A (LCA) from Myristica fragrans in non-small cell lung cancer cell lines—A549, NCI-H23, NCI-H520 and NCI-H460. LCA inhibited proliferation of all the four cell lines in a dose and time dependent manner with minimum IC50 of 20.03 ± 3.12, 22.19 ± 1.37 µM in NCI-H23 and A549 cells respectively. Hence NCI-H23 and A549 cells were used to assess the ability LCA to induce autophagy and apoptosis. LCA treatment caused G1 arrest, increase in Beclin 1, LC3II levels and degradation of p62 indicating activation of autophagy in both NCI-H23 and A549 cells. In addition, LCA mediated apoptotic cell death was confirmed by MMP loss, increased ROS, cleaved PARP and decreased pro-caspase3. To understand the role of LCA induced autophagy and its association with apoptosis, cells were analysed following treatment with a late autophagy inhibitor-chloroquine and also after Beclin 1 siRNA transfection. Data indicated that inhibition of autophagy resulted in reduced anti-proliferative as well as pro-apoptotic ability of LCA. These findings confirmed that LCA brought about autophagy dependent apoptosis in non-small cell lung cancer cells and hence it may serve as a potential drug candidate for non-small cell lung cancer therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the leading cancer worldwide and, among the different types of lung cancers non-small cell lung cancer (NSCLC) accounts for 80–85% of incidence [1]. Current treatment includes surgery, radiation therapy, chemotherapy and targeted therapy. Molecular characterization has led to progress in targeted cancer therapy in recent years. Mutations in epidermal growth factor receptor (EGFR) gene, RAS oncogene family gene (K-RAS) and anaplastic lymphoma kinase (ALK) gene are major marks for target based therapy. Several drugs and monoclonal antibodies as EGFR inhibitor, EGRF kinase inhibitor, ALK inhibitors have been discovered in recent years and are in clinical trials [2,3,4]. Despite the advances in treatment, cancer survival rate is about 15–18% only [5] and this is eventually due to drug resistance developed by cancer cells [6]. Poor prognosis and high mortality rate alarms us for early diagnosis and discovery of new therapeutic agents.
Apoptosis is a well-established type I programmed cell death mechanism and is the major route by which chemotherapy induces reduction in growth of cancer cells. However, many studies have reported that apoptosis is mitigated in certain cancer type and thus develop resistance to chemotherapy [7]. Therefore it is necessary to discover drugs that can cause cancer cell death by activation of other death inducing mechanism like autophagy.
Autophagy (self-eating) is a tightly regulated type II programmed cell death mechanism which involves four step process: (1) initiation of phagophore formation, (2) elongation of phagophore to engulf cells cytoplasmic contents like long-lived intracellular organelles and proteins which are sequestered into vacuoles known as autophagosome, (3) delivery or fusion of autophagosome with lysosome to form autophagolysosome followed by, (4) degradation of autophagosomal content by lysosomal enzymes and acids inside autophagolysosome, a process termed autophagic flux. The degrades are given back to cytoplasm to be reused for building macromolecules and thus helps in maintaining cellular homeostasis [8]. Autophagy is activated under starvation to provide nutrients required for cell survival. Autophagy in cancer has a dual role, tumor promotion and suppression [9]. Certain cancer type triggers autophagy to provide nutrients in order to overcome the chemotherapeutic stress [10,11,12]. However, prolonged expression of chemotherapy-induced autophagy leads to cancer cell death [13, 14].
Lignans derived from the seed and aril of Myristica fragrans (commonly known as nutmeg) have proven therapeutic values [15]. Licarin A(LCA), one such lignan has been reported for its anti-inflammatory, antiparasitic, anti-mycobacterial and anticancer properties [16,17,18,19]. Recent studies showed that lignans potentially activates autophagic cell death in many cancer cells [20, 21]. In the present study we isolated LCA from the seeds of nutmeg and evaluated its anti-proliferative potential, its ability to induce autophagy and apoptosis in non-small cell lung cancer cells lines. Further the association between autophagy and apoptosis was determined using a late autophagy inhibitor-chloroquine and gene knock down studies.
Materials and method
Reagent and chemicals
Nutmeg seeds were purchased from Kureekunnel Nutmeg Nursery and Gardens, Kottayam, Kerala, India. LCA isolated from seeds of nutmeg was dissolved in DMSO at a stock concentration of 20 mM and was stored in -80 °C. Cells treated with DMSO were considered as control. Dulbecco’s Modified Eagle’s Medium was purchased from Gibco. Fetal bovine serum and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were purchased from HiMedia. Chloroquine diphosphate, RIPA buffer, Protease inhibitor cocktail and Monodansylcadaverine were purchased from Sigma Aldrich. Beclin 1 (#3495), LC3A/B (#1274), p62 (#8025), pro-caspase 3 (#9665), PARP (#9542), caspase 3 (#9662), and p53 (#48818), anti-rabbit IgG, HRP-linked Antibody (#7074) were purchased from Cell Signalling. Cyclin D1 was purchased from Cloud clone (PAA585Hu01) and beta actin was purchased from Abcam (ab16039).
Isolation of Licarin A from nutmeg seeds
100 g of dried nutmeg seeds were ground and extracted twice with 75% methanol (400 ml, v/v) for 24 h at room temperature. Methanol extract was concentrated and fractionated with equal volume of n-hexane and ethyl acetate twice at room temperature. Ethyl acetate extract was concentrated and dried under reduced pressure. Further separation of ethyl acetate extract was performed using silica gel column chromatography (100–200 mesh size) and was eluted with n-hexane and ethyl acetate (10:1). 10 ml volume fractions were collected in each test tubes and HPTLC (60 F254, Merk) was performed simultaneously. Based on the HPTLC results, collected tubes were divided into Fraction 1–Fraction 4. Fraction 1 was further eluted with 20:1 n-hexane and ethyl acetate to obtain single compound C1 (crystalline, 35 mg). Fraction 2 was re-crystallized using petroleum ether and ethyl acetate (4:1) which led to isolation of a single compound C2 (colourless needle, 50 mg). Fraction 3 purification led to isolation C3 (white needle, 10 mg) and fraction 4 was highly viscous. NMR, Mass Spectrometry, FTIR, HPLC were performed to identify the compounds and to assess its purity of the compound.
Cell culture
Non-small cell lung cancer cell line, A549, NCI-H23, NCI-H460 and NCI-H520 were procured from National Centre for Cell Science, Pune, India. All cell lines were cultured in Dulbecco’s Modified Eagle’s Medium, supplemented with 10% (V/V) FBS in a humidified atmosphere at 37 °C with 5% CO2. Only the low passage number cells as received from the source were used to conduct experiments.
MTT assay
Cell viability was evaluated by MTT assay. Briefly 5 × 103 A549, NCI-H23, NCI-H460 and NCI-H520 cells/well were seeded in a 96-well plates and cultured for 24 h. Cells were then treated with various concentration ranging from 10 to 60 µM of LCA, control cells were treated with the vehicle (DMSO) and further incubated for 24 and 48 h. After the incubation period, MTT solution (0.5 mg/ml) was added to each well and incubated for another 4 h. Purple formazan crystal formed were dissolved in 100 µL DMSO and absorbance of each well was measured at a wavelength of 570 nm using SpectromaxM2e multimode reader.
Percentage inhibition was calculated by the using the formula:
where Ac and At are absorbance of DMSO control and test respectively.
CQ dose fixation
A549 and NCI-H23 Cell were cultured to about 70–80% confluence, treated with different concentration of CQ (30–150 µM) for 6, 9, 12 and 24 h and MTT was performed in after each indicated period. Also, Acidic vacuoles organelles (AVOs) formation was determined by acridine orange staining for A549 cells. Since 30 µM CQ mostly induced formation of Acidic vesicular organelles (AVOs) with minimal inhibition on A549 cells in 12 h, in the subsequent experiments, the dose of CQ was set at 30 µM, followed by a wash with PBS and treatment with indicated dose of LCA for another 24 h.
Cellular morphological changes
Briefly A549 and NCI-H23 cells were seeded in a 6 well plate (2 × 105 cells/well) and cultured for 24 h and treated with 25 µM of LCA for another 24 h and the cell morphology changes was observed using Optika inverted phase-contrast microscope.
Colony formation assay
A549 and NCI-H23 cells were cultured for 24 h in a 35 mm petridish at a density of 1000 cells per dish. Freshly prepared LCA at concentrations of 25 µM was added and incubated for 24 h at 37 °C with 5% CO2. Following incubation drug was removed; cells were washed with PBS and were cultured with drug-free complete medium for 12 days. Colonies formed stained with 1% methylene blue in 50% methanol. Colonies were counted manually.
Acidic vacuoles staining by acridine orange (AO)
Briefly A549 and NCI-H23 cells were seeded in a 24 well plate (5 × 104 cells/well), cultured for 24 h and treated with LCA for another 24 h at 37 °C with 5% CO2. After treatment cells were washed with PBS, stained with AO solution (10 µg/mL) and viewed under Optika fluorescence microscope with blue excitation wavelength for acidic vacuole formation characteristics.
Monodansylcadaverine (MDC) staining
MDC staining was performed to examine autophagic vacuole formation by LCA. Treated cells were incubated with 50 µM of MDC for 30 min at 37 °C. Following incubation cells were washed twice with PBS and observed using OPTIKA fluorescence microscope with UV excitation wavelength.
Acridine orange and ethidium bromide staining (AO/EB)
Acridine orange and Ethidium Bromide dual staining method was performed to examine apoptotic changes in treated cells. Cells were cultured in a 24 well plate and treated with LCA for 24 h. After treatment cells were washed with PBS, stained with AO/EB staining solution (10 µg/mL) and viewed using OPTIKA fluorescent microscope using blue excitation wavelength.
DAPI staining
Chromosome condensation and DNA fragmentation were observed by DAPI stating technique. Treated cells were fixed using 4% paraformaldehyde for 10 min and permeabilized with 70% ethanol for 5 min. Finally, cells were stained with DAPI (1 µg/ml) and viewed under Optika fluorescent microscope with UV wavelength.
Intracellular ROS measurement
After treatment with LCA cells were incubated with 10 µM of 2′,7′-dichlorofluorescein diacetate (DCF-DA) for 15 min at room temperature. Then cells were washed with PBS and imaged under Optika fluorescence microscope with blue excitation filter. Also, fluorescence intensity was measured using Guava 8HT Flow cytometer.
Detection of mitochondrial membrane potential
Mitochondrial membrane potential was evaluated by fluorescence microscopy and flow cytometry using Rhodamine-123 staining method. Treated cells were stained with 10 µM of Rho 123 for 15 min and observed under fluorescence microscope with blue excitation filter. DAPI was used for staining the nuclei.
Cells were stained with 10 µM of DAPI at room temperature for 45 min followed by PI for 10 min. Fluorescence intensity was measured using Guava 8HT Flow cytometer.
Cell cycle analysis
Briefly, 3 × 105 cells/well in 6 well plate and treated with LCA for 24 h. After incubation cells were trypsinized and washed with PBS. The cells were suspended in ice cold 70% ethanol for fixation at 4 °C overnight. Finally, cells were stained with propidium iodide/RNAse solution (50 µL of 100 µg/mL RNase and 200 µL of 50 µg/mL propidium iodide) and analysed using Guava 8HT Flow cytometer.
Immunofluorescence staining
Cells were grown on coverslip and treated with LCA for 24 h. CQ was used as positive control. After treatment, cells were fixed with 100% ice cold methanol at 20 °C for 15 min and then washed with PBS for 3 times, 5 min each. Following washing, cells were blocked with 5% BSA in 0.3% tween for 1 h and then incubated with primary antibody, LC3A/B (#1274, cell signalling, 1:100 dilution) at 4 °C overnight. After primary antibody incubation, cells were washed with PBS for 3 times 5 min each and incubated with secondary antibody (Alexa fluor 555, # A21428, Invitrogen, 1:500 dilutions) for 1 h in dark. Cells were further stained with DAPI for 5 min, washed with PBS for 3 times 5 min each and mounted on slide with prolong super gold Antifade reagent (#9071, cell signalling).Imaging was done using Confocal microscopy, LSM 700, Carl Zeiss.
Quantitative reverse-transcriptase PCR for mRNA expression
Gene expressions of autophagy markers were quantified using Quantitative reverse-transcriptase PCR (qRT-PCR). Total RNA was isolated as per trizol method and the RNA concentration was measured using nanodrop at 260 and 280 nm. Following RNA quantification, cDNA was synthesized by using High-Capacity cDNA Reverse Transcription Kit applied biosystems kit (Thermofisher scientific) according to manufacturer’s instruction. mRNA expression for Beclin 1 and LC3 were quantified using Lightcycler® 480 SYBR Green I Master kit (Roche applied science). GAPDH was used as the control housekeeping primer for each sample.
Primer details are as follows:
Gene | Forward | Reverse |
|---|---|---|
BECLIN 1 | CTGGACACTCAGCTCAACGTCA | CTCTAGTGCCAGCTCCTTTAGC |
MAP1LC3A | GCTACAAGGGTGAGAAGCAGCT | CTGGTTCACCAGCAGGAAGAAG |
GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA |
Small interfering RNA (siRNA) transfection
Cells were grown in a 6 well plate until 60% confluence and transfected with 100 nM SignalSilence® Control siRNA (Unconjugated) (#6568(−), cell signalling technology.) and 100 nM SignalSilence® Beclin-1 siRNA I (+) (#6222(+), cell signalling technology.) using Lipofectamine™ RNAiMAX transfection reagent (#13778-075, Life technologies.) following manufacturer protocol. After 48 h of transfection cell were treated with LCA for 24 h and protein levels of Beclin 1, LC3, p62, caspase 3 and PARP were detected using western blotting.
Western bolting
Cells were cultured in 100 mm plate at a density of 1 × 106 cells and treated with indicated concentration of LCA with or without CQ pre-treatment and CQ alone for 24 h. After treatment, the cytosolic proteins were extracted by lysing cells with 400 µL radio-immunoprecipitation assay (RIPA) buffer with constant agitation for 30 min at 4 °C followed by centrifugation of the lysate at 12,000 rpm for 15 min. The supernatant was carefully collected and protein concentration was measured by Bradford method using BSA as standard. 25 µg protein was resolved in a 10% SDS PAGE, transferred to PVDF membrane using semi-dry blot transfer (Bio-Rad) and blocked with 5% Skim milk or 5% BSA in TBS-Tween (0.01%) for 1 h at room temperature. After incubation, the membrane was washed thrice with TBS-Tween (0.01%) and further incubated with primary antibody at 4 °C overnight. After washing thrice with TBS-Tween (0.01%) membrane was incubated with anti-rabbit IgG, HRP-linked Antibody for 1 h at room temperature. Detection was performed using Enhanced chemiluminescence Substrate (ECL) (Thermo Scientific Pierce). Beta-actin was used as loading control. Band intensity of protein expression was quantified using the Image-J software. Band intensity of protein expression was quantified using Image-J software.
Statistical analysis
The results were analysed using Graphpad prism 5 software and Microsoft excel. The data is represented as mean ± SD of three individual experiments.
Results
Isolation and purification of Licarin A from Myristica fragrans (nutmeg)
Three compounds were isolated from ethyl acetate fraction of seeds of nutmeg and were assessed for antiproliferative effect against NSCLC cells using MTT assay. C1 showed no cytotoxic effect, C3 had solubility issue and C2 showed potent growth inhibition activity in NSCLC cells. Hence, C2 was used for further studies. To identify the structure and mass of the purified compound 2, NMR, FTIR and Mass spectrometry analysis was performed. Through detailed analysis of data, the compound purified was identified as Licarin A (LCA) with molecular weight of 326.16 from EI-MS 327.16 [M+1]+ (Fig. 1a), with reference to previously reports [17, 22]. Purity of the compound was assessed by analytical HPLC which showed a single peak at 254 nm.

Antiproliferative effect of LCA in non-small cell lung cancer cell. a Mass spectrometry analysis and the structure of the Licarin A isolated from Myristica fragrans (nutmeg). b–f A549, NCI-H23, NCI-H520 and NCI-H460 cells were treated with various concentrations of LCA ranging from 10 to 60 µM for 24 and 48 h and the percentage cell inhibition was calculated by comparing with the DMSO treated control cells using MTT assay, and the IC50 was calculated using GraphPad Prism 5 software. Data represented as mean ± SD of three independent experiments
Licarin A inhibits the proliferation of non-small cell lung cancer cell lines
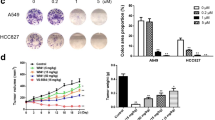
The growth inhibitory effect of LCA in non-small cell lung cancer cell lines: A549, NCI-H23, NCI-H520 and NCI-H460 were determined by MTT assay. Cells were treated with various concentrations of LCA ranging from 10 to 60 µM for 24 and 48 h. LCA was found to inhibit proliferation of A549, NCI-H520 and NCI-H460 cells in a dose and time dependent manner (Fig. 1b–e) with an IC50 of A549: 26.85 ± 2.769 and 22.19 ± 1.37 µM, NCI-H23: 26.39 ± 4.22 and 20.03 ± 3.12 µM, NCI-H520: 34.06 ± 3.06 and 30.2 ± 5.53 µM, and NCI-H460: 44.15 ± 3.21 and 35.01 ± 4.15 µM after 24 and 48 h respectively (Fig. 1f). Subsequent experiments were carried out with A549 and NCI-H23 cells considering the anti-proliferative activity of LCA with minimum IC50 in these cell lines. Cells when examined for morphological changes under phase contrast microscope following treatment with 25 µM of LCA showed elongated and distorted structure with large number of vacuoles compared to control cells (Fig. 2a).The effect of LCA on colony formation ability was also assessed. Results showed that LCA effectively decreased the colony forming ability of both A549 and NCI-H23 cells when compared to control cells (Fig. 2b, c).

Cellular morphological changes were observed using Phase contrast microscope. a A549 and NCI-H23 cells were treated with 25 µM of LCA for 24 h and change in the cell morphology was observed. Treated cells showed elongated and distorted structures with lager number of vacuoles compared to DMSO treated control cells. b, c The effect of LCA on colony formation was evaluated by treating cells with 25 µM of LCA for 24 h. After incubation drug containing media was replaced with fresh media and further cultured for 12 days. Colonies formed were stained using methylene blue and counted. Data represented as mean ± SD of three independent experiments. **P < 0.01, when compared with the control
Licarin A induced autophagy in non-small cell lung cancer cells
Autophagy is a self-degradation process in which the cell’s cytoplasmic content is degraded to provide nutrition under starvation and stress conditions, thus maintaining the cell homeostasis [23]. Autophagy in cancer cells has been reported to exhibit both pro-survival and pro-death role. Autophagy can be activated by the cancer cells to provide nutrients during chemotherapy that prolongs cancer cell survival. However, constitutive autophagy flux leads to cell death of the cancer cells [24]. In this study, presence of large number of vacuole structures observed in LCA treated cells prompted us to investigate whether autophagy is induced by LCA. Formation of Acidic vacuole organelles (AVOs) is a characteristic feature of autophagy. To observe AVOs, LCA treated cells were stained with acridine orange, a lysosomotropic dye which easily penetrate into cells, gets protonated in the acidic compartments, accumulates and emits characteristic orange red fluorescence when excited with blue excitation wavelength. In this study, LCA treated A549 and NCI-H23 cells showed increased number for orange red AVOs while it was rarely seen in control cells (Fig. 3a). Presence of autophagic vacuoles was further confirmed by staining cells with monodansylcadaverine, a dye that specifically stains autophagic vacuoles wherein LCA treated cells exhibited punctuated staining of the autophagic vacuoles while control cells showed diffused staining (Fig. 3a).

LCA induces autophagy in non-small cell lung cancer cells, A549 and NCI-H23. a Acidic vacuole organelles (AVOs) were observed by incubating cells with 10 µg/ml of acridine orange (AO) for 10 min and imaging was done using fluorescence microscope with blue excitation wavelength. Autophagic vacuoles were further confirmed by staining the cells with 50 µM of Monodansylcadaverine (MDC), a dye that specifically stains autophagic vacuoles for 30 min at RT and imaging them using fluorescence microscope with UV excitation wavelength. b, c mRNA expression of the autophagy markers Beclin 1 and LC3 II were quantified by Real Time PCR. d, Immunofluorescence staining to visualize LC3 localization. Cells were fixed with 100% ice cold methanol washed and incubated with specific primary antibody for LC3 protein and probed with a secondary antibody conjugated with alexa fluor 555 that emits red fluorescence when excited at 533 nm. Cellular nucleus was stained with DAPI and imaging was done using confocal microscopy (×20). Cells were data represented as mean ± SD of three independent experiments. *P < 0.05 and **P < 0.01 compared to control
In order to assess the level of autophagy markers in LCA treated cells, mRNA expression of Beclin-1, which is involved in autophagy initiation and microtubule-associated protein light chain 3 (LC3) involved in autophagy elongation were quantified using real time PCR. Exposure to LCA induced significant increase in mRNA levels of both Beclin-1 and LC3 at 10 and 20 µM concentrations after 24 h (Fig. 3b, c). These results indicated that LCA induced autophagy in A549 and NCI-H23 cells in a dose and time dependent manner.
In order to visualize localization of LC3, immunofluorescence staining was performed by using specific primary antibody for LC3 protein and a secondary antibody conjugated with alexa fluor 555 dye that emits red fluorescence while nucleus was stained with DAPI. Confocal microscopy images showed more punctuated localization of LC3 in the cytoplasm and nucleus of LCA treated cells when compared to control cells (Fig. 3d). All the above data confirmed that LCA induces autophagy in non-small cell lung cancer cells.
Effect of CQ on cell proliferation of non-small cell lung cancer cells
Chloroquine (CQ) is lysosomotropic weak base, which in the monoprotonated form diffuses into lysosomes, gets diprotonated and trapped. Protonated chloroquine increases lysosomal pH and thereby inhibit autophagosome degradation in lysosomes [25]. MTT assay (time and dose) and acridine orange staining for AVOs were performed to determine the dose at which CQ showed more AVOs formation with minimal growth inhibition. At 30 µM concentration and 12 h exposure, CQ showed weak cytotoxic activity with more AVOs formation while increased cytotoxicity was observed at 24 h for the same dose (Sup Fig. 1a, b). Based on these results, 30 µM of CQ for 12 h was selected as pre-treatment does and time for further experiments.
CQ pre-treatment reduced the cell death induced by LCA non-small cell lung cancer cells
To understand the role of autophagy in LCA mediated cell death, cells were pre-treated with 30 µM of chloroquine (CQ) for 12 h followed by incubation with various concentrations of LCA for 24 h and growth inhibition was determined by MTT assay (Fig. 4a). We found that CQ pre-treatment decreased the growth inhibitory effect of LCA. To support this finding, protein expression of autophagy markers such as Beclin-1, LC3 II (active form) and autophagy flux marker—Sequestosome-1/ubiquitin-binding protein (p62) were determined by western blot analysis in LCA treated cells with or without CQ pre-treatment. In LCA alone treated cells, a dose dependent increase in Beclin-1, LC3 II levels and augmented degradation of p62 was observed while in CQ alone treated cells increased levels of Beclin-1, LC3 II and accumulation of p62 was seen which indicated effective blockage of autophagy by CQ (Fig. 4b). Most importantly, the blockade effect of CQ in the degradation of p62 was found overcome by LCA (Fig. 4b–d), indicating enhanced autophagy flux. These findings revealed that LCA induced autophagy plays a death promoting role in non-small cell lung cancer cells.

CQ pre-treatment reduces the growth inhibitory effect of LCA in A549 and NCI-H23 cells. a Cells were treated with various doses of LCA (10–50 µM) with or without CQ 30 µM pre-treatment (12 h) for 24 h and the percentage cell growth inhibition was quantified using MTT assay. b Western blot analysis for the expression of the autophagy markers Beclin 1, LC3 II, p62 and β-actin (loading control) in cells lysate treated with different dose of LCA, CQ alone or CQ pre-treatment followed by LCA at the indicated doses for 24 h. c–e Graphical representation of the protein expression levels of Beclin 1, LC3 II, and p62 normalized with the loading control. Data represented as mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001, compared to control and #P < 0.05, ##P < 0.01 and ###P < 0.001 compared to LCA 20 µM
Effect of Licarin A on the cell cycle arrest in non-small cell lung cancer cells
We wanted to determine the effect of LCA on the cell cycle distribution. Cells were treated with 25 µM of LCA for 24 h and analysed using flow cytometer after staining them with PI/RNAse solution. LCA treatment caused increased accumulation of cells in the G1 phase when compared to control which suggested LCA induced G1 arrest (Fig. 5a).We further assessed the levels of tumor suppressor protein p53 which causes G1 cell cycle arrest triggered by DNA damage and cyclin D1 protein that plays a role in G1/S transition [26]. Protein expression of p53 increased while cyclin D1 levels decreased in LCA treated cells in a time dependent manner (Fig. 5b, c). These results indicated that LCA caused p53 dependent G1 arrest in A549 and NCI-H23 cells.

The effect of LCA on cell cycle distribution and intracellular ROS generation in A549and NCI-H23 cells. a Cells were treated with 25 µM of LCA for 24 h. The DNA was stained with Propidium iodide and cell cycle distribution was determined by flow cytometry. b, c Western blot analysis of protein levels of tumor suppressor protein p53 and cyclin D1 in cells lysate treated with 20 µM of LCA for indicated time period. Graphical representation of the protein expression levels normalized with the loading control. d Dichlorodihydrofluorescein diacetate (DCF-DA) staining was performed to examine the intracellular ROS generation by LCA. Cells were treated with 25 µM of LCA for 24 h, after incubation cells were stained with 10 µM of DCF-DA and imaging them using fluorescence microscope (blue excitation wavelength), also cells were subjected to flow cytometry analysis. Data represented as mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control
Effect of Licarin A in the generation of intracellular ROS in non-small cell lung cancer cells
Several studies have reported the role of intracellular reactive oxygen species (ROS) in the induction of autophagy and apoptosis [27, 28]. Therefore we wanted to check if LCA induces intracellular ROS generation in A549 and NCI-H23. Briefly, cells were treated with 25 µM LCA for 24 h and were stained with 2′,7′-dichlorofluorescein diacetate (DCF-DA), a cell permeable dye which is deacetylated by cellular esterase and is oxidized byROS into 2′,7′-dichlorofluorescein that emits green fluorescence [29]. LCA treated cells showed bright green fluorescence compared to control cells (Fig. 5d). The intensity of fluorescence was measured by flow cytometer as well (Fig. 5d).
Licarin A induced apoptotic cell death in non-small cell lung cancer cells
Having understood that LCA induces autophagy that plays a pro-death role, causes p53 dependent G1 arrest and increases intracellular ROS in A549and NCI-H23 cells, we further wanted to investigate the effect of LCA on apoptosis. Firstly, acridine orange and ethidium bromide staining was performed to examine apoptotic changes in LCA treated cells. Acridine orange is a vital dye that stains both live and dead cells. Ethidium bromide stains only those cells that have lost membrane integrity. Live cells appear uniformly green and early apoptotic cells stain green with bright green punctate in the nuclei due to chromatin condensation and nuclear fragmentation. Late apoptotic cells incorporate ethidium bromide and therefore stain orange, showing condensed and fragmented nuclei. Necrotic cells stain orange, but have a nuclear morphology resembling that of viable cells [30]. In the present study, cells treated with LCA showed early and few late apoptotic bodies and very few cells with necrotic changes (Fig. 6a).

LCA induced apoptosis in non-small cell lung cancer cell line A549 and NCI-H23. Cells were treated with 25 µM of LCA and DMSO control cells for 24 h and examined for the apoptotic changes using different staining techniques. a Acridine orange and Ethidium bromide staining for detection of early apoptotic, late apoptotic and necrotic bodies. DAPI staining for examination of DNA condensation. Rhodamine 123 and DAPI dual staining for evaluation of mitochondrial membrane potential. b Rhodamine 123 flow cytometry analysis for mitochondrial membrane potential. c, d Western blot analysis of protein levels of pro-caspase 3 in cells lysate treated with 20 µM of LCA for indicated time period. Graphical representation of the protein expression levels normalized with the loading control. Data represented as mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001, compared to control
DAPI staining technique is commonly used for identification of DNA condensation and nuclear fragmentation. DAPI preferentially binds to the minor groove of A-T rich region of the DNA [31]. In this study, treated cells showed bright and fragmented nuclei indicating that LCA caused both DNA condensation and nuclear fragmentation (Fig. 6a). Further, the effect of LCA on mitochondrial membrane potential (MMP) was assessed using Rhodamine 123 staining. MMP loss is associated with early events in the intrinsic apoptotic pathway. Rhodamine 123, a cationic dye gets accumulated in intact mitochondria and emits green fluorescence. Any loss in mitochondrial membrane potential leads to decrease in the intensity of Rhodamine 123 [32]. In this study, a remarkable reduction in the fluorescence intensity was observed in LCA treated cells as compared to control indicating loss of MMP (Fig. 6a). The same was confirmed by flow cytometry analysis (Fig. 6b). Also, protein level of pro-caspase 3 was found to be decrease following treatment with 20 µM of LCA for various time periods (Fig. 6c, d). These results confirmed that LCA activated apoptosis in non-small cell lung cancer cells A549 and NCI-H23.
Licarin A induced autophagy dependent apoptotic cell death in non-small cell lung cancer cells
The association of autophagy and apoptosis in LCA treated cells was investigated by assessing the protein expression levels of caspase 3 and PARP with or without CQ pre-treatment. Levels of pro-caspase3 were found decreased while cleaved PARP increased in LCA alone treated cells. On the other hand, in CQ pre-treated cells increased expression of pro-caspase 3 and decreased cleaved PARP were observed when compared to LCA alone treated cells indicating, autophagy plays a major role in LCA induced apoptosis in non-small cell lung cancer cells A549 and NCI-H23. (Fig. 7a, b).

Western blots analysis of apoptotic markers and siRNA transfection in A549 and NCI-H23. Cells were treated with different dose of LCA, CQ alone or in combination or CQ pre-treatment followed by LCA at the indicated doses for 24 h. Cells were lysed to isolate proteins and western blot analysis was performed, a, b Caspase 3 and PARP. Graphical representation of the protein expression levels normalized with the loading control. c Western blot analysis of protein from five group of A549 and NCI-H23 cells. group 1 treated with DMSO alone (lane 1); group 2 cells treated with LCA (20 µM); group 3 transfected with 100 nM SignalSilence® Control siRNA (Unconjugated); group 4 cells were transfected with 100 nM SignalSilence® Beclin-1 siRNA I (+) and group 5 cells were first transfected with 100 nM SignalSilence® Beclin-1 siRNA I for 48 h and treated with LCA (20 µM). Data represented as mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001, compared to control, #P < 0.05 and ##P < 0.001 compared to LCA 20 µM
Further to confirm the role of autophagy induced by LCA, we performed autophagy gene knockdown assay by silencing Beclin 1 gene using Beclin 1 siRNA along with western blot analysis of autophagy and apoptosis markers. Firstly, Beclin 1 levels in Beclin 1 siRNA transfected cells were significantly reduced when compared to control siRNA transfected cells confirming effective transfection. Further, in cells transfected with Beclin 1 siRNA followed by LCA treatment the levels of Beclin 1 and LC3II were found significantly reduced, while p62 levels were increased when compared to LCA alone treated cells indicating the induction of autophagy by LCA in A549 and NCI-H23 cells (Fig. 7c). Protein expression levels of cleaved caspase 3 and PARP were significantly decreased in Beclin 1 siRNA transfected cells followed by LCA treatment compared to the LCA alone treated cells thus confirming LCA induces autophagy mediated apoptotic cell death in non-small cell lung cancer cells (Fig. 7c).
Discussion
Phytochemicals are of interest for treatment of several diseases including cancer because of their low toxicity and less side effects. Previous reports showed that lignans isolated from Myristica fragrans have many therapeutic uses including anti-diabetic, anti-inflammatory, hepato protective, anticancer properties etc [16,17,18]. Licarin A isolated from Miliusa fragrans was previously reported for its anticancer activity in KB (oral carcinoma), MCF-7 (Breast cancer) and NCI-H187 (Small cell lung cancer) [18]. However, the effect of LCA on non-small cell cancer and the underlying mechanism has not yet been reported. In the present study, we isolated Licarin A from the seeds of Myristica fragrans (nutmeg) and demonstrated that the purified LCA exhibited antiproliferative effect in a dose and time dependent manner in non-small cell lung cancer cell line lines, A549, NCI-H23, NCI-H460 and NCI-H520 with lesser IC50 value in A549 and NCI-H23 cells.
Autophagy, a type II programmed cell death is an evolutionarily conserved process in which a cell self-eats its long lived or unfolded proteins and cytoplasmic organelles to maintain cellular homeostasis [33]. This process is tightly regulated by a distinct set of proteins named Atg (autophagy-related) [8]. Autophagy has been reported for its dual role in tumorigenesis. Cancer cells induce autophagy under stressful or in nutrition deprived conditions thus promoting cell survival [34], but constitutive or sustained autophagy leads to cancer cell death [23, 35]. A recent study reported that Honokiol, a lignan derived from bark of magnolia trees induced apoptosis and autophagy in K-ras mutant lung cancer cell lines [20]. In this study we observed LCA induced autophagy in A549 and NCI-H23 cells characterized by increased autophagic vacuoles, upregulation of mRNA levels of autophagy markers—Beclin 1 and LC3A as well as conversion of LC3 I to LC3II, an indicator of autophagosome formation along with degradation of p62, an autophagy flux indicator. Furthermore, inhibition of autophagy with chloroquine (lysosome inhibitor) remarkably decreased the cytotoxic effect of LCA by downregulating the degradation of p62 thereby suggesting that autophagy induced by LCA play a pro-death role in non-small cell lung cancer cells [20, 36].
Cell cycle is essential for cell proliferation which is a tightly regulated process with checkpoints in each phase and if cells failed to meet necessary needs in any phase, the progression of cell cycle is halted [37]. p53 controls cell proliferation mostly by intervening with the G1 phase of the cell cycle. p53 is activated as a result of DNA damage due to chemotherapy, radiation or hypoxia, increases the expression of p21 which in turn inhibits Cyclin D1 leading to G1 arrest [38]. In the present study, we observed that LCA increased p53 expression as well as caused G1 arrest as observed by flow cytometry. It is evident that following increased p53 levels and G1 cell cycle arrest, cells may enter apoptosis [39]. In this study we observed that cells treated with LCA exhibited increased intracellular ROS, collapse in the mitochondrial membrane potential, DNA condensation, downregulation of pro-caspase 3 and upregulation of cleaved PARP. These results pointed towards induction of apoptosis by LCA in A549 and NCI-H23 cells.
Several reports showed that apoptosis and autophagy are interlinked either positively or negatively [10, 40, 41]. Also many reports suggest that autophagy and apoptosis go hand in hand to cease cancer cell growth [20, 42]. To understand if autophagy induced by LCA play a cyto-protective or pro-death role, cells were pre-treated with autophagy inhibitor-chloroquine. We observed that inhibition of autophagy resulted in reduction of LCA mediated cell growth inhibition, decreased degradation of p62, upregulated pro-caspase3 and downregulated cleaved PARP levels. Similar results were observed in Beclin 1 gene silencing study also which confirmed that autophagy was an early event before induction of apoptosis. Findings from this study together reiterate that LCA induced autophagy-dependent apoptotic cell death in non-small cell lung cancer cells.
Conclusion
Ours is the first to report on the cytotoxic effect of LCA on non-small cell lung cancer cells which is brought about by G1 arrest, activation of autophagy that culminated in apoptotic cell death. Further, LCA increased the levels of p53 and intracellular ROS which could have contributed to the activation of autophagy and apoptosis (Fig. 8). However, further study to unravel the regulatory mechanism by which LCA induces autophagy needs to be carried out. Findings from this study throw light on the mode of action of LCA and hence it can serve as a potential drug candidate in future for non-small cell lung cancer therapeutics.

Schematic representation of the mechanism of LCA induced cell death in NSCLC. LCA exposed cells underwent apoptosis as well as autophagy. LCA upregulates autophagy markers Beclin-1 and activates LC3-II leading to autophagosome formation. Consequently, LCA activates p53 and ROS generation that would have induced autophagy and apoptosis. CQ pre-treatment inhibits autophagy which in turn reduces LCA induced cell death confirming that autophagy activated by LCA plays a pro-death role
Change history
15 March 2018
The original version of this article unfortunately contained a mistake.
Abbreviations
- LCA:
-
Licarin A
- CQ:
-
Chloroquine
- LC3:
-
Microtubule-associated protein 1 light chain
- EGFR:
-
Epidermal growth factor receptor
- p62:
-
Sequestosome-1/ubiquitin-binding protein
- p53:
-
Tumor suppressor protein
- ROS:
-
Reactive oxygen species
- DCFH-DA:
-
2′,7′-Dichlorofluorescein-diacetate
- PI:
-
Propidium iodide
- PARP:
-
Poly(ADP-ribose) polymerase
- NMR:
-
Nuclear magnetic resonance
- FTIR:
-
Fourier transform infrared
- HPLC:
-
High performance liquid chromatography
References
García-Campelo R, Bernabé R, Cobo M et al (2015) SEOM clinical guidelines for the treatment of non-small cell lung cancer (NSCLC) 2015. Clin Transl Oncol 17:1020–1029. https://doi.org/10.1007/s12094-015-1455-z
Liu SV, Zhao W, Petrini I et al (2017) Third-generation tyrosine kinase inhibitors targeting epidermal growth factor receptor mutations in non-small cell lung cancer. Front Oncol. https://doi.org/10.3389/fonc.2017.00113
Wang M, Wang G, Ma H, Shan B (2017) Crizotinib versus chemotherapy on ALK-positive NSCLC: a systematic review of efficacy and safety. Curr Cancer Drug Targets. https://doi.org/10.2174/1568009617666170623115846
Singh Malik P, Raina V (2012) Lung cancer: prevalent trends & emerging concepts. Int Agency Res Cancer Indian Counc Med Res 141:2009–2011. https://doi.org/10.4103/0971-5916.154479
Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS FEC SEER Cancer Statistics Review, 1975-2014 National Cancer Institute, Bethesda, MD, Based on November 2016 SEER data submission, posted to SEER web site, April 2017.
Housman G, Byler S, Heerboth S et al (2014) Drug resistance in cancer: an overview. Cancers 6:1769–1792. https://doi.org/10.3390/cancers6031769
Indran IR, Tufo G, Pervaiz S, Brenner C (2011) Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim Biophys Acta 1807:735–745. https://doi.org/10.1016/j.bbabio.2011.03.010
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221:3–12. https://doi.org/10.1002/path.2697.Autophagy
Morselli E, Galluzzi L, Kepp O et al (2009) Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta 1793:1524–1532. https://doi.org/10.1016/j.bbamcr.2009.01.006
Ishaq M, Khan MA, Sharma K et al (2014) Gambogic acid induced oxidative stress dependent caspase activation regulates both apoptosis and autophagy by targeting various key molecules (NF-κB, Beclin-1, p62 and NBR1) in human bladder cancer cells. Biochim Biophys Acta 1840:3374–3384. https://doi.org/10.1016/j.bbagen.2014.08.019
Peng X, Gong F, Chen Y et al (2014) Autophagy promotes paclitaxel resistance of cervical cancer cells: involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis 5:e1367. https://doi.org/10.1038/cddis.2014.297
Sharma N, Thomas S, Golden EB et al (2012) Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett 326:143–154. https://doi.org/10.1016/j.canlet.2012.07.029
Li LH, Wu P, Lee JY et al (2014) Hinokitiol induces DNA damage and autophagy followed by cell cycle arrest and senescence in gefitinib-resistant lung adenocarcinoma cells. PLoS ONE. https://doi.org/10.1371/journal.pone.0104203
Wang FZ, Fei HR, Cui YJ et al (2014) The checkpoint 1 kinase inhibitor LY2603618 induces cell cycle arrest, DNA damage response and autophagy in cancer cells. Apoptosis 19:1389–1398. https://doi.org/10.1007/s10495-014-1010-3
Jinous Asgarpanah (2012) Phytochemistry and pharmacologic properties of Myristica fragrans Hoyutt.: a review. Afr J Biotechnol 11:12787–12793. https://doi.org/10.5897/AJB12.1043
Ma CJ, Sung SH, Kim YC (2004) Neuroprotective lignans from the bark of Machilus thunbergii. Planta Med 70:79–80. https://doi.org/10.1055/s-2004-815463
Néris PLN, Caldas JPA, Rodrigues YKS et al (2013) Neolignan Licarin A presents effect against Leishmania (Leishmania) major associated with immunomodulation in vitro. Exp Parasitol 135:307–313. https://doi.org/10.1016/j.exppara.2013.07.007
Sawasdee K, Chaowasku T, Lipipun V et al (2013) New neolignans and a lignan from Miliusa fragrans, and their anti-herpetic and cytotoxic activities. Tetrahedron Lett 54:4259–4263. https://doi.org/10.1016/j.tetlet.2013.05.144
León-Díaz R, Meckes-Fischer M, Valdovinos-Martínez L et al (2013) Antitubercular activity and the subacute toxicity of (-)-licarin a in balb/c mice: a neolignan isolated from Aristolochia taliscana. Arch Med Res 44:99–104. https://doi.org/10.1016/j.arcmed.2012.12.006
Luo L, Li Y, Liu Z-Q et al (2017) Honokiol induces apoptosis, G1 arrest, and autophagy in KRAS mutant lung cancer cells. Front Pharmacol 8:199. https://doi.org/10.3389/fphar.2017.00199
Zhou J, Hu H, Long J et al (2013) Vitexin 6, a novel lignan, induces autophagy and apoptosis by activating the Jun N-terminal kinase pathway. Anticancer Drugs 24:928–936. https://doi.org/10.1097/CAD.0b013e328364e8d3
Nguyen PH, Kang HW, Le TVT et al (2011) Simple process for the decrease of myristicin content from Myristica fragrans (nutmeg) and its activity with amp-activated protein kinase (AMPK). J Food Biochem 35:1715–1722. https://doi.org/10.1111/j.1745-4514.2010.00496.x
Mah LY, Ryan KM (2012) Autophagy and cancer. Cold Spring Harb Perspect Biol 4:a008821. https://doi.org/10.1101/cshperspect.a008821
Brech A, Ahlquist T, Lothe RA, Stenmark H (2009) Autophagy in tumour suppression and promotion. Mol Oncol 3:366–375. https://doi.org/10.1016/j.molonc.2009.05.007
Harhaji-Trajkovic L, Arsikin K, Kravic-Stevovic T et al (2012) Chloroquine-mediated lysosomal dysfunction enhances the anticancer effect of nutrient deprivation. Pharm Res 29:2249–2263. https://doi.org/10.1007/s11095-012-0753-1
Giono LE, Manfredi JJ (2006) The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol 209:13–20. https://doi.org/10.1002/jcp.20689
Kongara S, Karantza V (2012) The interplay between autophagy and ROS in tumorigenesis. Front Oncol 2:171. https://doi.org/10.3389/fonc.2012.00171
Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M (2015) Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol 4:184–192. https://doi.org/10.1016/j.redox.2014.12.003
Eruslanov E, Kusmartsev S (2010) Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol 594:57–72. https://doi.org/10.1007/978-1-60761-411-1_4
Kasibhatla S (2006) Acridine orange/ethidium bromide (AO/EB) staining to detect apoptosis. Cold Spring Harb Protoc 2006:pdb.prot4493–prot4493. https://doi.org/10.1101/pdb.prot4493
Chazotte B (2011) Labeling nuclear DNA using DAPI. Cold Spring Harb Protoc 6:pdb.prot5556. https://doi.org/10.1101/pdb.prot5556
Baracca A, Sgarbi G, Solaini G, Lenaz G (2003) Rhodamine 123 as a probe of mitochondrial membrane potential: evaluation of proton flux through F0 during ATP synthesis. Biochim Biophys Acta 1606:137–146. https://doi.org/10.1016/S0005-2728(03)00110-5
Yang ZJ, Chee CE, Huang S, Sinicrope FA (2011) The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 10:1533–1541. https://doi.org/10.1158/1535-7163.MCT-11-0047
Guo XL, Li D, Sun K et al (2013) Inhibition of autophagy enhances anticancer effects of bevacizumab in hepatocarcinoma. J Mol Med 91:473–483. https://doi.org/10.1007/s00109-012-0966-0
Xiao D, Bommareddy A, Kim SH et al (2012) Benzyl isothiocyanate causes FoxO1-mediated autophagic death in human breast cancer cells. PLoS ONE 7:e32597. https://doi.org/10.1371/journal.pone.0032597
Wang Q, Chen Z, Diao X, Huang S (2011) Induction of autophagy-dependent apoptosis by the survivin suppressant YM155 in prostate cancer cells. Cancer Lett 302:29–36. https://doi.org/10.1016/j.canlet.2010.12.007
Kastan MB, Bartek J (2004) Cell-cycle checkpoints and cancer. Nature 432:316–323. https://doi.org/10.1038/nature03097
Vermeulen K, Berneman ZN, Van Bockstaele DR (2003) Cell cycle and apoptosis. Cell Prolif 36:165–175. https://doi.org/10.1046/j.1365-2184.2003.00267.x
Haupt S, Berger M, Goldberg Z, Haupt Y (2003) Apoptosis—the p53 network. J Cell Sci 116:4077–4085. https://doi.org/10.1242/jcs.00739
Chun J, Kang M, Kim YS (2014) A triterpenoid saponin from Adenophora triphylla var. japonica suppresses the growth of human gastric cancer cells via regulation of apoptosis and autophagy. Tumor Biol 35:12021–12030. https://doi.org/10.1007/s13277-014-2501-0
Li C, Chen J, Lu B et al (2014) Molecular switch role of Akt in Polygonatum odoratum lectin-induced apoptosis and autophagy in human non-small cell lung cancer A549 cells. PLoS ONE 9:1–12. https://doi.org/10.1371/journal.pone.0101526
Liu B, Cheng Y, Zhang B et al (2009) Polygonatum cyrtonema lectin induces apoptosis and autophagy in human melanoma A375 cells through a mitochondria-mediated ROS-p38-p53 pathway. Cancer Lett 275:54–60. https://doi.org/10.1016/j.canlet.2008.09.042
Acknowledgements
The research work was funded by Department of Biotechnology, Govt. of India, and DBT-IPLS program (BT/PR14554/INF/22/125/2010). We thank Department of Biochemistry and Molecular Biology for fluorescence microscope facility and Central Instrumentation Facility (CIF), Pondicherry University. We also thank Translational Research Platform for veterinary Biologicals-TANUVAS, Chennai for confocal microscopy facility.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
A correction to this article is available online at https://doi.org/10.1007/s10495-018-1452-0.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Maheswari, U., Ghosh, K. & Sadras, S.R. Licarin A induces cell death by activation of autophagy and apoptosis in non-small cell lung cancer cells. Apoptosis 23, 210–225 (2018). https://doi.org/10.1007/s10495-018-1449-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-018-1449-8