
Abstract
Apoptotic injury participates in hepatic fibrosis, but the molecular mechanisms are not well understood. The present study aimed to investigate the role of inducible TIMP1 in the pathogenesis of hepatic apoptosis-fibrosis. Apoptosis was induced with GCDC, LPS, and alcohol in precision-cut liver slices or bile duct ligation (BDL) in rats, as reflected by caspase-3 activity, TUNEL assay, and apoptosis-related gene profiles. The hepatic fibrosis was detected with Picrosirius staining, hydroxyproline determination, and expression profiling of fibrosis-related genes. Levels of TIMP1 were upregulated by the hepatic apoptosis, but downregulated by caspase inhibitor. The inducible TIMP1 was apoptosis-dependent. Once TIMP1 was inhibited with treatment of TIMP1-siRNA, the fibrotic response was reduced as demonstrated by hydroxyproline assay. In addition, the expression of fibrosis-related genes aSMA, CTGF, and TGFb2r were down-regulated subsequent to the treatment of TIMP1-siRNA. TIMP1 could mediate the expression of fibrosis-related genes. TIMP1 was transcriptionally regulated by nuclear factor c-Jun as demonstrated by EMSA and ChIP assay. The treatment of c-Jun siRNA could significantly decrease the expression of TIMP1 induced by alcohol, GCDC, or LPS treatment. Hepatic apoptosis induces the expression of TIMP1. Inducible TIMP1 can modulate the expression of fibrosis-related genes in liver. TIMP1 pathway is a potential target for therapeutic intervention of fibrotic liver diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Apoptosis is a ubiquitous feature in liver disease. Hepatic apoptosis can be caused by different inducers such as alcohol, pharmacological use of drugs, medicinal use of weight loss supplements, hepatotropic viruses, attacks by free fatty acids in the metabolic syndrome, and immune-mediated inflammatory processes afflicting the liver [1]. Apoptosis is generally considered a fast process without inflammatory response, since activation of caspases is quickly followed by cell fragmentation and phagocytosis [2]. Caspases are central initiators and executioners of the hepatic apoptosis [3, 4]. Caspase-3, a particular signaling caspase, is either partially or totally responsible for the proteolytic cleavage of many key proteins such as the nuclear enzyme poly (ADP-ribose) polymerase [5]. Chronic liver injury is characterized by frequent apoptosis, regeneration, and progressive fibrosis, despite different etiologies. The hepatocyte apoptosis is prominent in hepatic pathology, which is believed as a pivotal step in most forms of liver injury.
Tissue inhibitor of metalloproteinase-1 (TIMP1) is a natural glycoprotein that can inhibit the activation of matrix metalloproteinases (MMPs) [6]. The MMPs, a family of secreted proteolytic enzymes, function in the biosynthesis of connective tissue [7]. MMPs degrade constituents of the basal membrane and the extracellular matrix, including collagens, proteoglycans, gelatin, fibronectin, laminin, and elastin, under physiological and pathological conditions [8]. The MMPs can form complexes with TIMP inhibitors, which regulate the biological activities of the proteases [9]. TIMP1 is an essential member of TIMP family and vigorously participates in pathophysiologic activities. For example, down-regulation of tissue TIMP1 in aged human skin contributes to matrix degradation and impairs cell growth and survival [10]. TIMP1 may take part in matrix remodeling and involve endocarditis and degenerative valvular disease [11]. TIMP1 balances the role of MMPs in the central nervous system, which is associated with several inflammatory diseases [12]. TIMP1 protein has an antiapoptotic function and is able to promote cell proliferation in a wide range of cell types [13]. These mechanisms are important in a broad range of malignancies. TIMP1 is expressed in a subset of malignant lymphomas, which stimulates cell survival [14]. The TIMP1 expression is amplified in pancreatic cancer cells [15] as well as non-small cell lung cancer [16]. The augmented TIMP1 expression plays an adverse prognosis in the adenocarcinoma, as compared with the squamous cell carcinoma subtype [17]. TIMP1 is correlated with melanocyte malignant transformation and cancer cell survival [18]. TIMP1 also is a contributory factor in the development of liver fibrosis as proven in murine experimental models and patient samples [19]. Overexpression of TIMP1 itself did not induce liver fibrosis in TIMP1 transgenic mice under control of the albumin promoter/enhancer. After treatment with CCl4, however, TIMP1 had a significant increase in transgenic mice as compared with the control mice. TIMP1 does not singlehandedly induce liver fibrosis, but strongly exacerbates liver fibrosis progression. [20]. In a study on resolution of liver fibrosis, ribonuclease protection analysis demonstrated a rapid decrease in expression of the collagenase inhibitors TIMP1 and TIMP2, whereas collagenase mRNA expression remained at levels comparable to peak fibrosis [21]. In clinical patients with chronic hepatitis C, TIMP1 was one of serum markers in liver fibrosis [22]. TIMP1, mainly produced by activated hepatic stellate cells (HSCs) and Kupffer cells (KCs), plays a critical role in liver matrix remodeling.
Hepatic apoptosis has been implicated in the progression of fibrotic liver disease [23]. The mechanisms likely involve professional phagocytic cells engulfing apoptotic bodies such as CpG-DNA motifs or other mediators released by apoptotic cells. Engulfment of apoptotic bodies can activate a variety of signaling cascades, resulting in myofibroblasts (activated HSCs) that generate collagen 1 and the pro-fibrogenic cytokine TGF-β [24]. Inhibition of apoptosis with caspase inhibitors manifests the beneficial effects in murine models of hepatic fibrosis [25]. Apoptosis resistance is associated with TIMP1 expression in different cell types as well. Liver injury has shown differential levels of TIMP1 expression, but the relationship between hepatic apoptosis and TIMP1 remains obscure. Mechanistic links are still undetermined. The present study investigates the pathophysiologic role of TIMP1 subsequent to apoptotic liver injury. By analyzing TIMP1 expression with fibrosis-related gene expression profiles, we provide novel insight into the role of apoptosis-TIMP1 network in fibrotic liver diseases. These results refine the molecular mechanisms, through which apoptosis mediates the process of liver fibrosis. The comprehensive role of TIMP1 may be an interesting target for therapeutic intervention of liver disease.
Materials and methods
Precision-cut liver slice (PCLS) culture
Wistar rat liver was perfused with cold-Hanks’ balanced salt solution supplemented with 5 mM glucose and 50 μg/ml gentamycin. The consecutive PCLS (8 mm diameter, 250 μm thickness) were incubated for 2 h in Williams’ E medium enforced with 0.35 μM insulin, 0.1 μM dexamethasone, and 5 % fetal calf serum (FCS) to promote adherence to the mesh [26]. Then, PCLS were treated for 28 h with 30 μM of GCDC, 1.5 μg/ml of LPS, and 40 mM of alcohol, respectively [27–29]. Regular culture of PCLS was performed with Williams’ E medium containing insulin, dexamethasone, and 1 % FCS. In each experiment, at least six PCLS per condition were used.
Cell culture
Hepatocytes were isolated from adult Wistar rat liver along standard liver perfusion procedure. Primary hepatocyte culture was described previously [30].
Cell co-culture
HSCs were co-cultured with KCs as previously described [31]. Briefly, primary cells were isolated by liver perfusion with bacterial pronase and liberase, followed by density gradient centrifugation with Histodenz (11 % over 17.5 %) and elutriation to separate KCs from endothelial cells. Purity of the HSC fraction (∼95 %) was assessed by autofluorescence, and that of the KC fraction (∼96 %) by immunolabeling with the monoclonal antibody RPE-ED2. A ratio of KC:HSC was maintained at 3:1. After 1 day incubation, both KCs and HSCs were washed triple times with serum-free DMEM/F12 and cell-culture inserts containing the KCs were transferred onto the HSCs. New medium (4 ml without serum) was added to HSCs cultured with KCs or with empty inserts as control.
Animal procedures
Wistar rats received humane care according to the criteria outlined by the National Academy of Sciences and the National Institutes of Health. The common bile duct was ligated through a ventral laparotomy [32].
RNA interference
The double stranded siRNA against TIMP1 or c-Jun (GenBank accession number: NM_053819.1 for TIMP1; NM_021835.3 for c-Jun) were consisted of pools of three target-specific 19–25nt siRNA in accordance with Ambion web-based criteria. Double-stranded siRNA (0.1 mg/kg) was injected through rat portal vein 20 h ago, prior of sacrifice for PCLS preparation [33].
Caspase assay
Liver tissue was homogenized with RIPA lysis buffer (Sigma-Aldrich). 100 μg of lysate protein and colorimetric substrate IETD-pNA were utilized for caspase assay. The activity was calculated as pmol/min [2].
TUNEL Assay
The TdT-FragEL™ DNA fragmentation detection kit was obtained from Calbiochem [2]. Apoptotic rate is calculated by counting the number of TUNEL-positive cells in 100× microscopic fields.
Western blotting assay
Protein samples were resolved with 10 % SDS–polyacrylamide gel electrophoresis and blotted with appropriate primary antibodies at dilution of 1: 500–1,000. Horseradish peroxidase-conjugated signal was detected using ECL chemiluminescent substrate (Amersham Biosciences).
Quantitative real-time PCR (qRT-PCR)
Total tissue RNA was extracted with TRI Reagent (Molecular Research Center, Cincinnati, OH). cDNA was synthesized using cDNA Synthesis Kit (Invitrogen). The level of gene expression was calculated by a mathematical delta–delta method, which was normalized to the expression level of 18S rRNA.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared with the modified Dignam protocol [2]. The consensus binding was labeled with digoxin. Protein–DNA complexes were separated from the unbound DNA probe through 6 % native polyacrylamide gels.
Chromatin immunoprecipitation (ChIP) Assay
As previously described [30], the crude nuclear extract was re-suspended in Lysis Buffer and then gotten sonication at a power setting of 30 %. 4 μg of rabbit antiserum was used for the immunoprecipitation. Crosslinks were reversed by 400 μl of Elution Buffer. 1 μl of DNA was amplified with suitable primers that covered the potential binding area.
Statistical analysis
All results represented experiments from a minimum of three separate preparations. Data were expressed as mean ± SD unless otherwise indicated. Comparisons were performed using Student’s t test or two-way analysis of variance followed by Bonferroni correction where appropriate. A p value of ≤0.05 was considered to be statistically significant.
Results
TIMP1 was upregulated subsequent to apoptosis
In vitro apoptosis was induced in precision-cut liver slices (PCLS) by different inducers such as GCDC, LPS, and alcohol, respectively. Viabilities were indicated by MTT assay (Supplement Fig. A). The in vivo apoptosis was caused by common bile duct ligation (BDL) in rats. Apoptosis, reflected by caspase-3 activity (Supplement Fig. B, C) and TUNEL assay (Supplement Fig. D–G), resulted in an upregulation of TIMP1 expression at mRNA level through qRT-PCR (Fig. 1a, b) and protein detection by Western blotting (Fig. 1c). TIMP1 levels were increased in BDL livers, as compared with the sham livers. Time-course analysis revealed that the TIMP1 expression was correlated to caspase-3 activity following BDL treatment (Fig. 2a, b). Taken together, the apoptotic liver injury could upregulate the expression of TIMP1 as demonstrated by qRT-PCR and Western blotting. These causative factors might have different mechanisms to initiate apoptosis, but TIMP1 expression was significantly elevated.

Expression of TIMP1 was increased by apoptosis-inducers. a expression of TIMP1 at mRNA levels through qRT-PCR. Precision-cut liver slices (PCLS) was treated for 28 h with 30 μM of GCDC, 1.5 μg/ml of LPS, and 40 mM of alcohol, respectively. Histograms represent mean ± SD of 7–10 PCLS per group. **P < 0.01 versus control. b mRNA levels of liver TIMP1 from control (or sham, n = 3) or BDL (n = 5) treatment for 2 weeks. c protein levels of TIMP1 by Western blotting

Expression of TIMP1 was correlated with apoptosis. a levels of TIMP1 and caspase-3 over time in BDL animals. Results represent mean ± SD of 4–6 rats at each timepoint. b the correlative analysis of TIMP1 and caspase-3 activity (P = 0.0025)
Upregulation of TIMP1 was apoptosis-dependent
TIMP1 expression was markedly enhanced by different apoptosis-inducers. It was thus hypothesized that the upregulation of TIMP1 might depend on the apoptotic status. To test this hypothesis, precision-cut liver slices were treated with pan-caspase inhibitor Z-VAD-FMK (50 μM) [34] and then challenged with the aforementioned GCDC, LPS, and alcohol, respectively. The Z-VAD-FMK suppressed caspase activation and decreased apoptosis as demonstrated by caspase-3 activity and TUNEL assay (Fig. 3a, b). When apoptosis was inhibited in PCLS, TIMP1 expression was decreased as determined by the mRNA level of TIMP1 (Fig. 3c). TIMP1 protein showed a similar trend to the level of gene expression (Fig. 3d; Supplement Fig. H). While liver slices were stimulated with insulin-like growth factor 1 (IGF-1), levels of TIMP1 were not altered by the IGF-1 treatment (Supplement Fig. I). These results suggest that the inducible TIMP1 is mediated by apoptotic process or apoptosis-dependent, not through growth signal pathway.

Upregulation of TIMP1 was apoptosis-dependent. a caspase-3 activity in PCLS, following apoptotic induction with or without 50 μM of pancaspase inhibitor Z-VAD-FMK. The PCLS were pre-incubated with Z-VAD-FMK for 30 min prior to GCDC, LPS, and alcohol treatment. *P < 0.05; **P < 0.01. b TUNEL assay after apoptosis-inducer treatment with or without Z-VAD-FMK. c, d TIMP1 expression was affected by Z-VAD-FMK at mRNA level through qRT-PCR as well as protein level via Western blotting
TIMP1 modulated the expression of fibrosis-related genes
Hepatic fibrosis was time-dependent as exhibited by Sirius staining (Fig. 4a; Supplement Fig. J, K) and hydroxyproline determination (Fig. 4b) in BDL rats. The hepatic fibrosis was also characterized by the expression profile of fibrosis-related genes composed of aSMA, Col1a1, CTGF, TGFβ1, TGFb2r, and TIMP1 (Fig. 4c, d). The ample expression of TIMP1 was positively correlated with accumulative levels of CTGF and TGFb2r (Fig. 4e, f). When TIMP1 was inhibited by TIMP1-siRNA through portal vein injection, the response of the tissue was investigated by qRT-PCR and Western blotting (Fig. 5a, b). To exclude the side effect of TIMP1 knockdown in hepatocytes, the effect of siRNA-mediated knockdown on hepatocyte apoptosis was examined. Hepatocytes were isolated from control or siRNA-treated livers following previous protocol (2). Primary hepatocytes were then stimulated with GCDC, LPS, and alcohol, respectively. Hepatocyte apoptosis was reflected by caspase-3 activity (Fig. 5c). The fibrotic response was quantitated by hydroxyproline assay (Fig. 5d). Interestingly, expression profiles of fibrosis-related genes aSMA, CTGF, and TGFb2r were significantly altered (Fig. 5e). Surely, the mechanism by which TIMP1 affects the expression of fibrosis-related genes is worthy of further investigation. In addition, the proliferating ability was detected by MTT assay and cyclin D1 level (Supplement Fig. L, M). Inflammatory cytokines TGFβ1 and TNFα were measured subsequent to TIMP1 knockdown as well (Supplemental Data, Fig. N). The modification of TIMP1 expression could modulate the hepatic fibrosis through the regulation of fibrosis-related gene expression and cell proliferation.

Apoptotic injury stimulated hepatic fibrosis. a quantitation of Sirius staining by NIH-published quantitative imaging software. Histograms represent mean ± SD of 4-6 rats at each timepoint. *P < 0.05; **P < 0.01. b hydroxyproline determination in BDL livers. c expression profile of fibrosis-related genes by qRT-PCR in PCLS. *P < 0.05; **P < 0.01. d expression profile of fibrosis-related genes in livers of BDL treatment for 2 weeks. e the expression of TIMP1 was positively correlated with accumulative level of CTGF (P = 0.0038). f the expression of TIMP1 was positively correlated with accumulative level of TGFb2r (P = 0.0224)

TIMP1 modulated the expression of fibrosis-related genes. a, b after TIMP1-siRNA treatment, the expression of TIMP1 was determined by qRT-PCR and Western blotting. c following TIMP1-siRNA treatment, primary hepatocytes were isolated and hepatocyte apoptosis was then induced by GCDC, LPS, and alcohol respectively. Caspase-3 activity was measured after treatment for 32 h. d the fibrotic response was determined by hydroxyproline assay. e expression profiles of fibrosis-related genes aSMA, CTGF, and TGFb2r. *P < 0.05; **P < 0.01
Expression of TIMP1 was detected in hepatocytes
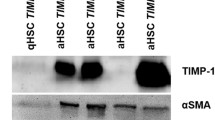
Apoptotic injury altered the level of TIMP1 in PCLS. In order to understand the inter-relationship between apoptosis and TIMP1 expression, it was necessary to know which cells were involved and how TIMP1 expression was activated in response to apoptotic injury. As we know, PCLS contains several phenotypically distinct cell types, e.g. hepatocytes, stellate cells, Kupffer cells, cholangiocyte, sinusoidal endothelial cells, etc. Predominant hepatocytes make up 70 % of the liver cells. Hepatocytes were thus isolated according to the standard procedure and the primary culture was carried out (2). Primary hepatocytes were targeted by ethanol, GCDC, and LPS toxicity. Hepatocyte apoptosis was revealed by caspase-3 activity (Fig. 6a). Levels of TIMP1 in hepatocytes were analyzed through real-time PCR, which were compared with that in PCLS (Fig. 6b). The levels of TIMP1 in hepatocytes did not show any significant difference between apoptotic treatment and control group. The source of inducible TIMP1 might be independent of the hepatocytes. Additionally, no difference was found for hydroxyproline measurement (Fig. 6c). To address the producer cells of inducible TIMP1, levels of TIMP1, CTGF, and αSMA were analyzed by real-time PCR after primary stellate cells and Kupffer cells were co-cultured with apoptotic bodies generated by hepatocytes (Fig. 6d) [31]. Expression levels of TIMP1 were also estimated in primary hepatocytes and stellate cells isolated from the BDL livers (Fig. 6e).

The relationship between apoptosis and TIMP1 in primary hepatocytes. a Hepatocyte apoptosis was detected by caspase-3 activity. b Levels of TIMP1 in hepatocytes were compared with that in PCLS via real-time PCR. c the measurement of hydroxyproline. d Levels of TIMP1, CTGF, and αSMA by real-time PCR after primary stellate cells and Kupffer cells co-cultured with apoptotic bodies generated by hepatocytes. e Expression levels of TIMP1 in primary hepatocytes and stellate cells isolated from the BDL livers. *P < 0.05; **P < 0.01
TIMP1 was regulated by nuclear factor c-Jun
TIMP1 gene promoter contains potential nuclear factor binding sites such as PPAR-γ1, Ets, STATs, SRE, SP1, NF-kb, c-Jun, c-Fos, HNF-1, HNF-4, HNF-6, etc. Some binding sites Ets, STATs, SRE, SP1 and LBP1, had been investigated [35], but other potential binding sites still need to be confirmed. Activation of c-Jun was altered in stellate cells and Kupffer cells co-cultured with apoptotic bodies generated by hepatocytes (Supplemental Data, Fig. O). The present study thus focused on the function of c-Jun nuclear protein in the regulation of TIMP1 expression. c-Jun combines c-Fos to form an AP-1 early response transcription factor. The inducible transcriptional complex AP-1 is crucial for cell adaptation to environmental changes [36]. A similarity of putative c-Jun binding sites is contrasted within −2.6 kb area of TIMP1 promoter. c-Jun binding sequences ttactcagcc and tgctgactcaggt are completely conservative in mouse, rat, and human genomes (Fig. 7a). EMSA was performed to examine whether c-Jun nuclear protein could bind TIMP1 promoter regions. Oligonucleotide probes containing the identical sequences for c-Jun binding sites were designed and optimized (Supplement Table 1). Results showed that in vitro-synthesized probes could bind nuclear protein (Fig. 7b). When c-Jun antibody was added into the complex of probe/nuclear protein, the binding signal was further inspected with gel supershift assay (Fig. 7c). EMSA and gel supershift assay demonstrated that there were c-Jun-specific DNA sequences in TIMP1 promoter. Next, ChIP assay was carried out to determine the ability of nuclear protein binding to DNA structure in native chromatin context. The crude nuclear extract was mechanically sheared by sonication to generate small protein-DNA fragments. These fragments were immunoprecipitated with c-Jun specific antibody or rat IgG as non-specific control (Fig. 7d). After having reversed the cross-links and removed proteins from the immunoprecipitated protein-DNA complexes, the purified DNA was analyzed by appropriate primers to cover potential binding regions (Supplement Table 2). The relative ratios of c-Jun specific antibody/IgG could reflect the result of ChIP assay (Fig. 7e). The ratio of c-Jun/IgG was significantly 15.5-fold (P = 0.0012). According to data from EMSA, gel supershift, and ChIP assay, we concluded that TIMP1 was regulated by transcription factor c-Jun. Subsequently, c-Jun was silenced by siRNA to investigate the role of c-Jun in regulation of TIMP1 expression. c-Jun siRNA was a pool of target-specific 19–25nt siRNAs designed to knock down c-Jun expression. The pretreatment of c-Jun siRNA could inhibit the expression of TIMP1 as detected by Western blotting (Fig. 7f). When c-Jun siRNA-pretreated PCLS were then challenged with alcohol, GCDC, and LPS, the expression of TIMP1 at mRNA level was significantly downregulated (Fig. 7g–i). Other fibrosis-related genes CTGF, TGFb2r were measured as well (Supplemental Data, Fig. P). Moreover, variation of c-Jun was assessed in primary hepatocytes and stellate cells separated from the liver of BDL-operated rats (Supplemental Data, Fig. Q).

TIMP1 expression was regulated by nuclear factor c-Jun. a potential c-Jun binding sequences ttactcagcc and tgctgactcaggt are completely conservative in mouse, rat, and human TIMP1 promoter within −2.6 kb area. b In vitro-synthesized probes could bind nuclear protein as demonstrated by EMSA. c gel supershift assay. d the crude nuclear extract was precipitated with cJun-specific antibody, while rat IgG as control. e the relative ratios of cJun-specific antibody/IgG could reflect the result of ChIP assay (P = 0.0012). f TIMP1 expression was inhibited by c-Jun siRNA through portal vein injection as detected by Western blotting. g–i after livers were pretreated by c-Jun siRNA, mRNA levels of TIMP1 in PCLS were altered by alcohol, GCDC, and LPS treatment. **P < 0.01
Discussion
Apoptotic injury was induced in precision-cut liver slices by GCDC, LPS, and alcohol or in rats through common bile duct ligation. Apoptosis could consistently upregulate the level of TIMP1 in spite of different initiating mechanisms. The molecular basis behind this phenomenon remains unknown. The upregulated TIMP1 was suppressed by caspase inhibitor. This suggests that the certain relationship is existed between apoptosis and TIMP1 expression. Furthermore, a modification of the TIMP1 expression altered expression profiling of fibrosis-related genes. Hepatic apoptosis was associated with fibrosis through the TIMP1-mediated pathway. Due to complexity of regulatory networks, it is unsure whether the TIMP1 signaling is the only pathway that regulates the progression of hepatic fibrosis. The treatment of c-Jun siRNA significantly decreased the expression of TIMP1. This is a novel approach to adjust the expression of TIMP1 at the transcriptional level. The blockage of c-Jun expression or TIMP1 pathway can be alternative approaches to interfere in the exacerbation of liver fibrosis.
Apoptosis increases TIMP1 expression. As rats were treated with alcohol intake and choline-deficient diet, this combined treatment triggered an apoptotic response as determined by elevated Bax, cytochrome c release, and caspase-3 activity. The combined treatment also stimulates a profibrogenic response due to upregulation of TIMP1, COL1α1, αSMA, as well as downregulation of MMP13, MMP2, and MMP9 [37]. Hepatitis C virus infection leads to hepatocyte apoptosis, which can activate HSCs and amplify the fibrogenic genes TIMP1, TIMP2, COL1α1, and TGFβ1 [38]. Apoptosis stimulates the expression of TIMP1. The upregulated TIMP1 would, in turn, inhibit apoptosis. TIMP1 expression could inhibit MMPs and was sufficient to block radiation-induced apoptosis in the capillary endothelial cells. The protective mechanism depended on the relative levels of MMPs and TIMPs [39]. TIMP1 is associated with apoptosis resistance that is related to negative prognosis in some human cancers. An overexpressed TIMP1 in melanoma cells reduced latency time for tumor appearance and increased metastatic potential. The elevated TIMP1 was combined with a progressive gene demethylation, which conferred anoikis resistance. TIMP1 may be a valued marker for melanocyte malignant transformation [18]. The TIMP1 expression inhibited apoptosis and mediated the survival of breast epithelial cells [40]. When apoptosis is induced during liver disease, a series of response is followed, e.g. activation of Kupffer cells and production of TIMP1. Phagocytic cells engulf apoptotic bodies such as CpG-DNA motifs [23, 24], which can release mediators to accelerate TIMP1 expression. TIMP1 further activates HSCs and synthesis of collagen, in mediation of the liver-fibrotic pathogenesis. In progressive fibrosis, HSCs proliferate and secrete collagen into the extracellular space. When the degradation of collagen accomplished by metalloproteinases is reduced by tissue inhibitors TIMPs, extracellular collagen is accumulated as demonstrated in liver and pulmonary fibrosis [19, 41]. TIMP1 plays an essential role in matrix remodeling. This is a dynamic process to connect apoptosis with fibrosis in liver diseases. Actually, the upregulation of TIMP1 can be understood as a stress response that is induced by apoptosis.
Progression of liver fibrosis is a wound healing response attributable to liver injury. During hepatic fibrosis, the activated HSCs undergo continuous proliferation as reflected by activation markers α-SMA. The excessive matrix proteins are secreted by HSCs. Expression of MMP1 is inhibited, whereas TIMP1 expression is upregulated. An imbalance between MMP1 and TIMP1 results in a net increase in extracellular matrix accumulation [42]. TIMP1 siRNA significantly reduced HSC proliferation. The attenuated proliferation was associated with reduced Akt phosphorylation and was partially rescued by addition of recombinant TIMP1. TIMP1 has a novel autocrine mitogenic effect on HSC, which may involve Akt-dependent and specific nuclear mechanisms of action [43]. Moreover, our data indicated that the treatment of TIMP1-siRNA could alter the profile of fibrogenic aSMA, CTGF, TGFβ1, and TGFb2r. In animals of spontaneous recovery from liver fibrosis, nuclease protection assay demonstrated a rapid decrease in expression of the collagenase inhibitors TIMPs. There was a reduction in the number of activated HSC. Apoptosis of the activated HSCs might vitally contribute to resolution of fibrosis [21]. Following treatment with CCl4 in TIMP1-transgenic mice, an active form of MMP2 level was decreased. The TIMP1 promotes the hepatic fibrosis by inhibiting both matrix degradation and apoptosis of HSCs [20]. An elevation of TIMP1 expression exacerbates the liver fibrosis through transgene, but inhibition of TIMP1 expression alleviates the fibrosis via siRNA silencing [20, 42]. Moreover, the fibrogenic role of TIMP1 expression is inducer-dependent. When rat liver fibrosis was induced with dexamethasone and CCl4 respectively, the correlation was different between TIMP1 expression and liver fibrosis in the two models. Serum TIMP1 level could positively correlate with the severity of liver fibrosis in immune (dexamethasone)-induced rats, but not in CCl4-induced experimental model [44]. The mechanisms through which TIMP1 enhances the liver fibrosis may include that TIMP1 can (i) stimulate the proliferation of HSCs; (ii) decrease the apoptosis of HSCs; (iii) inhibit MMPs; (iv) indirectly activate fibrosis-related gene expression or inhibit antifibrotic gene expression. Of note, TIMP1 knockout mice could develop more liver fibrosis than wild-type mice after CCl4 exposure [45]. TIMP-1 might have dual roles in liver fibrosis. The precise mechanism needs to be determined.
The regulatory mechanism of TIMP1 expression remains unknown. Hepatic apoptosis induces the expression of TIMP1 that is further associated with the fibrosis-related gene expression. The whole process is controlled by diverse aspects, including hepatic proliferation, matrix remodelling, and maintenance of hepatic phenotype. TIMP1 is highly inducible at the transcriptional level in response to many cytokines and hormones. c-Jun as a basic transcriptional molecule regulates TIMP1 expression. The relationship between c-Jun and hepatocyte apoptosis plays a critical role in the progression of liver dysfunction [46]. Hepatocyte apoptosis activates c-Jun expression that stimulates transcription of TIMP1 in Kupffer and stellate cells. TIMP1 antagonizes the c-Jun-mediated death signals. c-Jun has multiple functions. In the embryonic stage, c-Jun regulates organogenesis and cell differentiation via cell cycle progression and apoptosis. Postnatal hepatocyte proliferation and liver regeneration were impaired in mice lacking c-Jun in the liver [47]. c-Jun/AP-1 regulates liver regeneration through a molecular pathway that involves p53, p21, and the stress kinase p38α [36]. c-Jun can antagonize the proapoptotic activity of p53 and affect the initiation and promotion of liver tumor development [48]. The mechanism through which c-Jun regulates TIMP1 is still under investigation. c-Jun is an important component of AP-1 transcription factor complexes. AP-1, Ets-1, PEA3, and UTE-1 transcription factors are able to control TIMP1 promoter through their functional interactions [36, 37].
Fibrosis is a featured process in liver pathology. The progressive fibrosis enters the final stage or cirrhosis. The function is seriously harmed in cirrhotic livers. Currently, no satisfactory treatment is available in clinical practice. In recent years, the role of hepatic apoptosis has been recognized, especially in inducing models. The apoptosis-induced fibrosis can be roughly classified into two stages. The initial stage includes the release of cytokines and chemokines, which triggers an apoptotic response. Thereafter, the secondary stage reflects interactions of injured cells, Kupffer cells, HSCs, and extracellular matrix. The secondary response covers multiple aspects such as apoptosis-induced TIMP1 expression, proliferation, and regeneration. TIMP1 is positively correlated to immune-induced liver fibrosis in experimental rats [44]. The clinical significance of TIMP1 is its bridging role to connect apoptosis with liver fibrosis. A blockage of TIMP1 expression can interrupt an association of apoptosis with fibrosis, which may have a specific significance in cholestatic liver disorders. In summary, hepatic apoptosis stimulates TIMP1 expression. Levels of the inducible TIMP1 are apoptosis-dependent, which modulates the profile of fibrosis-related genes. TIMP1 is a major regulator of extracellular matrix. An overexpressed TIMP1 exacerbates liver fibrosis. Hepatic apoptosis may mediate fibrosis through TIMP1 signaling pathway. The current study also demonstrated that c-Jun regulated the expression of TIMP1. c-Jun/TIMP1 pathway has potential value in the treatment of fibrotic liver disease.
Abbreviations
- PCLS:
-
Precision-cut liver slices
- TIMP1:
-
Tissue inhibitor of metalloproteinase-1
- GCDC:
-
Glycochenodeoxycholate
- LPS:
-
Lipopolysaccharide
- EtOH:
-
Alcohol
- BDL:
-
Bile duct ligation
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- EMSA:
-
Electrophoretic mobility shift assay
- ChIP assay:
-
Chromatin immunoprecipitation assay
References
Malhi H, Guicciardi ME, Gores GJ (2010) Hepatocyte death: a clear and present danger. Physiol Rev 90(3):1165–1194
Wang K, Brems JJ, Gamelli RL, Ding J (2005) Reversibility of caspase activation and its role during glycochenodeoxycholate-induced hepatocyte apoptosis. J Biol Chem 280(25):23490–23495
Chandler JM, Cohen GM, MacFarlane M (1998) Different subcellular distribution of caspase-3 and caspase-7 following Fas-induced apoptosis in mouse liver. J Biol Chem 273:10815–10818
Leist M, Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2(8):589–598
Simbulan-Rosenthal CM, Rosenthal DS, Iyer S, Boulares AH, Smulson ME (1998) Transient poly(ADP-ribosyl)ation of nuclear proteins and role of poly(ADP-ribose) polymerase in the early stages of apoptosis. J Biol Chem 273(22):13703–13712
Khokha R, Waterhouse P (1994) The role of tissue inhibitor of metalloproteinase-1 in specific aspects of cancer progression and reproduction. J Neurooncol 18(2):123–127
McIntush EW, Smith MF (1998) Matrix metalloproteinases and tissue inhibitors of metalloproteinases in ovarian function. Rev Reprod 3(1):23–30
Sluijter JP, de Kleijn DP, Pasterkamp G (2006) Vascular remodeling and protease inhibition—bench to bedside. Cardiovasc Res 69(3):595–603
Brew K, Dinakarpandian D, Nagase H (2000) Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta 1477(1–2):267–283
Hornebeck W (2003) Down-regulation of tissue inhibitor of matrix metalloprotease-1 (TIMP-1) in aged human skin contributes to matrix degradation and impaired cell growth and survival. Pathol Biol (Paris) 51(10):569–573
Soini Y, Satta J, Määttä M, Autio-Harmainen H (2001) Expression of MMP2, MMP9, MT1-MMP, TIMP1, and TIMP2 mRNA in valvular lesions of the heart. J Pathol 194(2):225–231
Gardner J, Ghorpade A (2003) Tissue inhibitor of metalloproteinase (TIMP)-1: the TIMPed balance of matrix metalloproteinases in the central nervous system. J Neurosci Res 74(6):801–806
Offenberg H, Brünner N, Mansilla F, Orntoft Torben F, Birkenkamp-Demtroder K (2008) TIMP-1 expression in human colorectal cancer is associated with TGF-B1, LOXL2, INHBA1, TNF-AIP6 and TIMP-2 transcript profiles. Mol Oncol 2(3):233–240
Lai R, Rassidakis GZ, Medeiros LJ, Ramdas L, Goy AH, Cutler C, Fujio Y, Kunisada K, Amin HM, Gilles F (2004) Signal transducer and activator of transcription-3 activation contributes to high tissue inhibitor of metalloproteinase-1 expression in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Am J Pathol 164(6):2251–2258
Bramhall SR, Stamp GW, Dunn J, Lemoine NR, Neoptolemos JP (1996) Expression of collagenase (MMP2), stromelysin (MMP3) and tissue inhibitor of the metalloproteinases (TIMP1) in pancreatic and ampullary disease. Br J Cancer 73(8):972–978
Fong KM, Kida Y, Zimmerman PV, Smith PJ (1996) TIMP1 and adverse prognosis in non-small cell lung cancer. Clin Cancer Res 2(8):1369–1372
Simi L, Andreani M, Davini F, Janni A, Pazzagli M, Serio M, Orlando C (2004) Simultaneous measurement of MMP9 and TIMP1 mRNA in human non small cell lung cancers by multiplex real time RT-PCR. Lung Cancer 45(2):171–179
Ricca TI, Liang G, Suenaga AP, Han SW, Jones PA, Jasiulionis MG (2009) Tissue inhibitor of metalloproteinase 1 expression associated with gene demethylation confers anoikis resistance in early phases of melanocyte malignant transformation. Transl Oncol 2(4):329–340
Iredale J (2008) Defining therapeutic targets for liver fibrosis: exploiting the biology of inflammation and repair. Pharmacol Res 58(2):129–136
Yoshiji H, Kuriyama S, Miyamoto Y, Thorgeirsson UP, Gomez DE, Kawata M, Yoshii J, Ikenaka Y, Noguchi R, Tsujinoue H, Nakatani T, Thorgeirsson SS, Fukui H (2000) Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology 32(6):1248–1254
Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, Hovell C, Arthur MJ (1998) Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 102(3):538–549
Leroy V, Monier F, Bottari S, Trocme C, Sturm N, Hilleret MN, Morel F, Zarski JP (2004) Circulating matrix metalloproteinases 1, 2, 9 and their inhibitors TIMP-1 and TIMP-2 as serum markers of liver fibrosis in patients with chronic hepatitis C: comparison with PIIINP and hyaluronic acid. Am J Gastroenterol 99(2):271–279
Guicciardi ME, Gores GJ (2010) Apoptosis as a mechanism for liver disease progression. Semin Liver Dis 30(4):402–410
Torok NJ (2007) Apoptotic cell death takes its toll. Hepatology 46(5):1323–1325
Witek RP, Stone WC, Karaca FG, Syn WK, Pereira TA, Agboola KM, Omenetti A, Jung Y, Teaberry V, Choi SS, Guy CD, Pollard J, Charlton P, Diehl AM (2009) Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 50(5):1421–1430
Olinga P, Merema M, Hof IH, de Jong KP, Slooff MJ, Meijer DK, Groothuis GM (1998) Effect of human liver source on the functionality of isolated hepatocytes and liver slices. Drug Metab Dispos 26:5–11
Clouzeau-Girard H, Guyot C, Combe C, Moronvalle-Halley V, Housset C, Lamireau T, Rosenbaum J, Desmoulière A (2006) Effects of bile acids on biliary epithelial cell proliferation and portal fibroblast activation using rat liver slices. Lab Invest 86(3):275–285
Elferink MG, Olinga P, Draaisma AL, Merema MT, Faber KN, Slooff MJ, Meijer DK, Groothuis GM (2004) LPS-induced downregulation of MRP2 and BSEP in human liver is due to a posttranscriptional process. Am J Physiol Gastrointest Liver Physiol 287(5):1008–1016
Sawyer JS, Daller JA, Brendel K, Yohem K, Putnam CW (1994) The hepatotoxicities of endotoxin and ethanol comparisons in vitro using the precision-cut rat liver slice model. Life Sci 55(18):1407–1417
Wang K, Brems JJ, Gamelli RL, Holterman AX (2010) Survivin signaling is regulated through nuclear factor-kappa B pathway during glycochenodeoxycholate-induced hepatocyte apoptosis. Biochim Biophys Acta 1803(12):1368–1375
Nieto N (2006) Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology 44(6):1487–1501
Arias M, Sauer-Lehnen S, Treptau J, Janoschek N, Theuerkauf I, Buettner R, Gressner AM, Weiskirchen R (2003) Adenoviral expression of a transforming growth factor-beta1 antisense mRNA is effective in preventing liver fibrosis in bile-duct ligated rats. BMC Gastroenterol 3:29
Li G, Xie Q, Shi Y, Li D, Zhang M, Jiang S, Zhou H, Lu H, Jin Y (2006) Inhibition of connective tissue growth factor by siRNA prevents liver fibrosis in rats. J Gene Med 8(7):889–900
Vanhulle VP, Neyrinck AM, Pycke JM, Horsmans Y, Delzenne NM (2006) Role of apoptotic signaling pathway in metabolic disturbances occurring in liver tissue after cryopreservation: study on rat precision-cut liver slices. Life Sci 78(14):1570–1577
Bahr MJ, Vincent KJ, Arthur MJ, Fowler AV, Smart DE, Wright MC, Clark IM, Benyon RC, Iredale JP, Mann DA (1999) Control of the tissue inhibitor of metalloproteinases-1 promoter in culture-activated rat hepatic stellate cells: regulation by activator protein-1 DNA binding proteins. Hepatology 29(3):839–848
Stepniak E, Ricci R, Eferl R, Sumara G, Sumara I, Rath M, Hui L, Wagner EF (2006) c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38MAPK activity. Genes Dev 20:2306–2314
Nieto N, Rojkind M (2007) Repeated whiskey binges promote liver injury in rats fed a cholinedeficient diet. J Hepatol 46(2):330–339
Gieseler RK, Marquitan G, Schlattjan M, Sowa JP, Bechmann LP, Timm J, Roggendorf M, Gerken G, Friedman SL, Canbay A (2011) Hepatocyte apoptotic bodies encasing nonstructural HCV proteins amplify hepatic stellate cell activation: implications for chronic hepatitis C. J Viral Hepat 18(11):760–767
Vorotnikova E, Tries M, Braunhut S (2004) Retinoids and TIMP1 prevent radiation-induced apoptosis of capillary endothelial cells. Radiat Res 161(2):174–184
Li GY, Fridman R, Kim HRC (1999) Tissue inhibitor of metalloproteinase-1 inhibits apoptosis of human breast epithelial cells. Cancer Res 59:6267–6275
Swiderski RE, Dencoff JE, Floerchinger CS, Shapiro SD, Hunninghake GW (1998) Differential expression of extracellular matrix remodeling genes in a murine model of bleomycin-induced pulmonary fibrosis. Am J Pathol 152(3):821–828
Venugopal SK, Jiang J, Kim TH, Li Y, Wang SS, Torok NJ, Wu J, Zern MA (2010) Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am J Physiol Gastrointest Liver Physiol 298(1):101–106
Fowell AJ, Collins JE, Duncombe DR, Pickering JA, Rosenberg WM, Benyon RC (2011) Silencing tissue inhibitors of metalloproteinases (TIMPs) with short interfering RNA reveals a role for TIMP-1 in hepatic stellate cell proliferation. Biochem Biophys Res Commun 407(2):277–282
Nie QH, Zhang YF, Xie YM, Luo XD, Shao B, Li J, Zhou YX (2006) Correlation between TIMP-1 expression and liver fibrosis in two rat liver fibrosis models. World J Gastroenterol 12(19):3044–3049
Wang H, Lafdil F, Wang L, Yin S, Feng D, Gao B (2011) Tissue inhibitor of metalloproteinase 1 (TIMP-1) deficiency exacerbates carbon tetrachloride-induced liver injury and fibrosis in mice: involvement of hepatocyte STAT3 in TIMP-1 production. Cell Biosci 1(1):14
Schrum LW, Black D, Iimuro Y, Rippe RA, Brenner DA, Behrns KE (2000) c-Jun does not mediate hepatocyte apoptosis following NFkappaB inhibition and partial hepatectomy. J Surg Res 88(2):142–149
Behrens A, Sibilia M, David JP, Möhle-Steinlein U, Tronche F, Schütz G, Wagner EF (2002) Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. EMBO J 21(7):1782–1790
Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, Wagner EF (2003) Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell 112(2):181–192
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Wang, K., Lin, B., Brems, J.J. et al. Hepatic apoptosis can modulate liver fibrosis through TIMP1 pathway. Apoptosis 18, 566–577 (2013). https://doi.org/10.1007/s10495-013-0827-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-013-0827-5