
Abstract
Various mechanical stresses can induce apoptosis of nucleus pulposus (NP) cells and intervertebral disc (IVD) degeneration in vivo, but the underlying molecular mechanism by which the number of NP cells is decreased in degenerated IVD is still not elucidated. The purpose of this study was to investigate whether the mitochondrial pathway is involved in compression-induced apoptosis of rabbit NP cells. The compression apparatus was used to investigate the effect of the compression in this process at one magnitude (1.0 MPa) for 6, 12, 18, 24 and 36 h. Cell viability was measured by cell counting kit-8. Apoptosis rate was analyzed by flow cytometry and the morphologic changes in apoptosis cells were observed by the phase-contrast microscopy and Hoechst 33258 staining. The apoptosis-related gene and protein synthesis, such as Bax, Bcl-2 and Caspase-3, was analyzed by real-time polymerase chain reaction and Western-blot, respectively. Mitochondrial function was evaluated by analyzing the mitochondrial permeability transition pore (MPTP), as well as reactive oxygen species (ROS) and mitochondrial membrane potential (MMP). The results indicated that compression at the magnitude of all time points induced apoptosis of rabbit NP cells in a time-dependent manner, and the cell viability was reduced significantly. Furthermore, the compression at this level profoundly suppressed the functions of the mitochondria such as the opening of MPTP, the excessive production of ROS and the decreased MMP. Our findings suggest that the compression-induced IVD degeneration is mediated, at least in part, via the mitochondrial apoptotic pathway in NP cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Degeneration of intervertebral disc (IVD) is a primary cause of low back pain (LBP) and is a prerequisite to IVD hernia [1], which has a high social and economic cost. Notwithstanding this importance, the pathological mechanism of the IVD degeneration has not been fully defined. It’s generally believed that the degeneration of IVD is influenced by numerous factors such as age, genetics and mechanical stimulus in which the latter plays an important role in degeneration of IVD [2–6]. Although mechanical stress is an important modulator of the degeneration, the underlying molecular mechanism by which nucleus pulposus (NP) cells play roles in the degeneration of IVD is still not totally clear.
The IVD is composed of two conspicuous and interdependent anatomic structure: the surrounding annulus fibrosus (AF) and the central gelatinous NP. IVD cells especially NP cells play a very important role in maintaining the integrity of the IVD through producing collagen of type II, aggrecan and other ingredients which involved in extracellular-matrix (ECM) metabolism. The reduction of NP cells population and the loss of ECM are the central features in the aged and degenerated IVD. Some evidence reveals that the NP may be associated with aging and the initiation of IVD degeneration [7–9]. Many scholars suggest that the NP cells possess distinct characteristics necessary for IVD homeostasis, so this has stimulated some recent studies focused on the feature of NP cells [10]. Most studies in vitro and in vivo suggest that the cellular loss due to excessive apoptosis of disc cells plays an important role in the development of IVD degeneration [11, 12].
In the signaling pathways of apoptosis, there are two main caspase-dependent pathways, intrinsic and extrinsic, which are mediated by mitochondria and death-receptor, respectively. The extrinsic pathway is chiefly activated by Fas-receptor, in which the activated caspase-8 as an apical caspase directly activates effector caspases such as caspase-3 and then leads to cell apoptosis [13, 14]. The intrinsic pathway, also known as the mitochondria pathway, is another important pathway in the process of apoptosis [15, 16].
In addition to the well-defined role of mitochondria in energy metabolism, regulation of cell apoptosis has recently emerged as second major function of these organelles [17–22]. Mitochondrion has an important role in caspase-dependant and caspase-independent pathways of apoptosis. The intrinsic and extrinsic apoptotic pathways are regulated by various mitochondrial proteins. The mitochondria pathway which mainly activated by various cellular stresses and numerous apoptotic signals is another important pathway in the process of apoptosis. Cellular stress response or apoptotic signals can cause the release of mitochondrial cytochrome c, second mitochondria-activator of caspase (Smac), apoptosis inducing factor (AIF) and endonuclease (Endo G). Cytochrome c is combined with apoptotic protease activating factor-1 (Aparf-1), procaspase-9 and ATP to form apoptosome. And then it induces the activation of caspase-3 to trigger a cascade of caspases, leading to apoptosis. In addition, the release of AIF can cause nuclear condensation and the break of chromatin (Fig. 1).

Schematic diagram of mechanisms for mitochondria-mediated apoptotic pathway
Currently, a large number of studies found that the apoptotic mechanism of disc cells is associated with many factors, such as Fas ligand [23, 24]. However, the mitochondrial pathway has not been documented regarding NP cells apoptosis induced by mechanical stresses so far. Therefore, we investigated the role of mitochondrial pathway in the process of apoptosis induced by compression through detecting the apoptosis and the mitochondrial function.
Materials and methods
The isolation and culture of primary NP cells
The study was conducted under the protocol approved by the animal experimentation committee of Huazhong University of Science and Technology. The skeletally mature Japanese white rabbits (3 months, male) for this study were purchased from the Experiment Animal Center of Tongji Medical College, Huazhong University of Science and Technology, China. The animals were euthanized by air embolism. Thoracolumbar spines were removed under the aseptic condition. The surrounding soft tissues were completely removed to expose the disc. Each disc was cut transversely, and the gelatinous NP was separated from disc using a scalpel and spatula. Care was taken not to injury the underlying endplate. The obtained NP tissue was digested with 0.2% collagenase type II (Gibco, USA) for 30 min, filtered through a 70-μm cell strainer and washed three times with phosphate-buffered saline (PBS), and the suspension was centrifuged twice at 300×g for 5 min. The isolated cells were seeded in 6-well culture plates in Dulbecco’s modified Eagle’s medium/ham’s F-12 (DMEM/F-12, Gibco, USA) containing 20% fetal bovine serum (FBS, Gibco, USA) supplemented with 50 μg/mL ascorbate (Sigma, USA), 2 mM/L l-glutamine (Sigma, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (Beyotime, China) at 37°C, 5% CO2 atmosphere. Culture medium was changed every 2–3 days. Cells were passaged by 0.25% trypsinization (Beyotime, China) when they reached confluence of 80–90%. NP cells at the second generation maintained in a monolayer were used throughout the following experiments.
Application of a compression apparatus on NP cells
The cells were divided into six groups: control group (0 h), and five pressure groups, in which the cells were exposed to 1.0 MPa pressure for 6, 12, 18, 24, and 36 h, respectively. The control group samples were incubated at 37°C without any compression under the same culture conditions. According to the classic air-pressure principle, a custom-made compression apparatus (Fig. 2) in vitro was applied as previously described to expose NP cells to continuous high pressure [25]. The stainless pressure vessel allows for pumping compressed gas into it to form a closed high-pressure environment which is monitored with a barometer. The cell culture plates were put on the bottom of the pressure vessels which were filled with little distilled water to keep moisture. The pressure vessels were placed in an incubator at 37°C, and the digital thermometer was used to monitor the change of internal temperature. The mixed 0.5% CO2 + 99.5% compressed air was pumped into the pressure vessel, and the valve of the pumping ended when the pressure value reached 1.0 MPa.

Schematic illustration of custom-made compression apparatus 1 air-inlet/outlet, 2 thermometer, 3 barometer, 4 safety valve, and 5 bracket
Measurement of cell proliferation
The NP cells were seeded in 96-well culture plates at a density of 2 × 103 cells per well. Then the cells at different time points were exposed to the pressure after they have been cultured for 48 h. 20 μL cell counting kit-8 (CCK-8) solution was applied to each well, and incubated for 4 h at 37°C according to the manufacturer’s instructions. Cell proliferation, also the activity of mitochondrial dehydrogenase was assessed by absorbance detection at 450 nm with a microplate reader (Biotek, USA).
Detection of apoptosis by flow cytometry and confocal microscopy
The NP cells of second generation were placed in 6-well culture plates at 1 × 105 cells per well and treated with the same pattern. The apoptosis rate of NP cells was detected by Annexin V/PI (KeyGen Biotech, China) double staining according to the manufacturer’s instructions and as described previously [12, 26]. Briefly, the cells of different groups were collected by trypsinization and centrifugation, and then washed with ice-cold PBS twice and resuspended in 500 μL binding buffer. 5 μL fluorescein-conjugated Annexin V and 5 μL PI were added and further incubated in the dark for 15 min at room temperature. The apoptosis rate was analyzed by flow cytometry (BD LSR II, Becton Dickinson) using FACSDiva Software (Becton Dickinson, USA). The cells staining positive for annexin V and negative for propidium iodide and those positive for double staining were identified as apoptotic cells in each sample. They were counted and represented as a percentage of the total cell populations.
Cells were cultured, treated, collected and stained as stated above. Lastly, the stained cells suspension was observed under a confocal microscope (Nikon A1, Japan).
Observation of cell morphology (Hoechest 33258 staining)
Briefly, cells (5 × 104 cells per well) were seeded into a 6-well culture plate and cultured as described above. After treatment with compression, the medium was collected and the cells were harvested by trypsinization and centrifugation immediately. The cells were washed twice with PBS and then stained with 0.5 μg/mL Hoechst 33258 (Sigma, USA) for 15 min in the dark. Morphologic changes in apoptotic nuclei were observed and photographed under the fluorescence microscope (Olympus IX71) with emission wavelength at 460 nm and excitation wavelength at 350 nm.
Quantitative real-time polymerase chain reaction (RT-PCR) analysis for the gene expression of Caspase-3, Bax, Bcl-2
Total RNA was extracted from NP cells of each sample using 1 mL Trizol reagent (Invitrogen, USA) following the manufacturer’s instructions. The equal amounts (11 μL) of isolated RNA were transcribed into complementary DNA (Invitrogen, USA). Specific primer pairs (2.5 μM) were designed as follows: Bax: 5′-CGAGGTCTTTTTCCGAGTGG-3′, 5′-GCCCATAATAGTCCTGATGAGC-3′; Bcl-2: 5′-GCTACGAGTGGGATACTGGAGA-3′, 5′-GTAGCGACGAGAGAAGTCATCC-3′; Caspase-3: 5′-GCAAATCAATGGACTCTGGGA-3′, 5′-CATCACCGTGGCTTAGAATCA-3′; GAPDH: 5′-CGCCTGGAGAAAGCTGCTA-3, 5′-ACGACCTGGTCCTCGGTGTA-3′. The quantitative RT-PCR was performed by using a standard PCR kit of SYBR Green mix (ToYobo, Japan) and 2.5 μL cDNA in SLAN Real-Time PCR System (Shanghai Hongshi Medical Technology Co., China). The amplified products were subjected to the analysis of melting curve, and all of the data were analyzed by the method of 2-ΔΔCT calculation and standardized by the house-keeping gene GAPDH.
Western-blot analysis
Cells of different groups (0, 6, 12, 18, 24, and 36 h) were harvested and lysed in a standard buffer (Beyotime, China), and total protein was extracted using a Western and IP Cell Lysis Kit (Beyotime, China). The cell lysate was collected by centrifugation at 12,000×g for 10 min at 4°C. The protein concentrations were determined by using an enhanced BCA Protein assay kit (Beyotime, China). Equal amounts of protein from each sample were loaded on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel and transferred to the nitrocellulose membranes. Using nonfat milk to block the nonspecific blinding, then the membranes were incubated overnight at 4°C with rabbit polyclonal antibody against Bax (Ptglab, 1:1000), Bcl-2 (Ptglab, 1:500), Caspase-3 (Abcam, 1:500) and GAPDH (Abcam, 1:3000). After several times of washing and conjugation with secondary antibodies (KPL, 1:3000) which were labeled with horseradish peroxidase, the immunoreactive bands were visualized by the system of enhanced chemiluminescence detection (PerkinElmer, USA). The band intensity was quantified by the software of AlphaEaseFC 4.0.
Mitochondrial permeability transition pore (MPTP)
Cells (5 × 104 cells per well) seeded in 6-well culture plates, were treated with the compression as described above. MPTP was measured by MPTP Fluorescence Assay Kit (Genmed, China) according to the manufacturer’s instructions. The cells were collected by centrifugation at 300×g for 5 min, then 500 μL preheated cleaning solution (Reagent A) and equal amounts of staining working solution which contained staining solution (Reagent B) and neutralization solution were added to the cell suspension, respectively. Next they were mixed gently and incubated in the dark at 37°C for 20 min. At last the samples were resuspended in Reagent A again and detected by using fluorescence spectrophotometer (PerkinElmer LS55, USA).
Reactive oxygen species (ROS)
The intracellular ROS level was measured by ROS-specific fluorescent probe, 2-,7-dichlorofluorescin diacetate (DCFH-DA; Beyotime, China) [27]. The collected NP cells at a density of 2 × 105 cells/mL were resuspended in DCFH-DA and incubated in the dark at 37°C for 20 min. Then they were mixed every 3–5 min to make the probe contact with the cells fully and the cells were washed with serum-free culture medium three times to remove the probes which haven’t entered into the cells. The mean fluorescence intensity (MFI) of DCF in different samples was analyzed by using flow cytometry with an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
Observation of mitochondrial membrane potential (MMP) by flow cytometry
MMP was determined by the 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolycarbocyanine iodide (JC-1, Beyotime, China) staining following the manufacturer’s instructions. Briefly, the harvested NP cells were resuspended in the mixture of 500 μL culture medium and 500 μL JC-1 staining fluid, and then incubated in the dark at 37°C for 20 min. After washing with ice-cold staining buffer twice by centrifugation, cells were resuspended in 500 μL culture medium and analyzed by flow cytometry. The values of MMP staining from each sample were expressed as ratio of red fluorescence intensity over green fluorescence intensity.
Evaluation of MMP in situ
The NP cells were seeded into sterile Petri dish (diameter of 35 mm) special for the detection of laser scan confocal microscopy, at a density of 2 × 104 cells per well and then cultured for 48 h. After the treatment of pressure on NP cells at each time point, the cells were incubated with the mixture of 500 μL JC-1 staining fluid and 500 μL cell culture medium in the dark at 37°C for 30 min. Subsequently, the cells were washed twice with the staining buffer preserved in 4°C. Lastly, 1 mL cell culture medium was added into the each specimen and the cells were observed under the laser scan confocal microscope.
Statistical analysis
All of the experiments were repeated at least three times. Experimental values were obtained from three independent experiments with a similar pattern and expressed as means ± standard deviation (SD). Statistical analyses were performed using SPSS software package 12.0. Data were analyzed by one-way analysis of variance (ANOVA) followed by least significant difference (LSD) for comparison between control and treatment groups. For the assays of caspase-3 gene expression and apoptosis rate, data were compared using one-way ANOVA followed by Dunnett’s T3 test. Significance was set at P < 0.05.
Results
Compression inhibits the proliferation and the viability of NP cells in vitro
To examine the regulatory effect of compression pressure on the growth of NP cells, CCK-8 assay was used to determine cell proliferation by the changes in the activity of mitochondrial dehydrogenase. As shown in Fig. 3, the compression inhibited the proliferation of the NP cells of pressure groups in a definitive time-dependent manner. The P values of each treatment group were less than 0.001. There were statistically significant differences between control and treatment groups.

The proliferation capacity of NP cells measured by using CCK-8 assay. The value of CCK-8 could reflect the activity of mitochondrial dehydrogenase. Rabbit NP cells were subjected to the pressure of 1.0 MPa for indicated time points (6, 12, 18, 24 and 36 h, respectively), and the group of 0 h served as control. Data are expressed as mean ± SD from three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001 vs. control; ANOVA/LSD test)
Compression induces apoptosis of NP cells
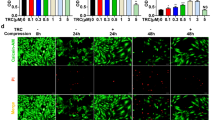
The effect of compression on apoptosis of NP cells was examined by flow cytometry by Annexin V/PI dual staining. Flow cytometry revealed that compression induced apoptosis of NP cells in a time-dependent manner. As shown in Fig. 4a and b, an evident elevation of apoptosis rate at different time points was noted following the treatment of compression. But there was no statistically significant difference in the elevated apoptosis rate between 6-h group and control group. P values of each treatment group were 0.062, 0.003, 0.004, 0.025 and 0.011, respectively. Furthermore, the early apoptotic cells accounted for the majority proportion from Fig. 4a and this phenomenon was also demonstrated by Fig. 4c.

Compression-induced apoptosis of NP cells in a time-dependent manner. a Representative dot plot of cell apoptosis by flow cytometry analysis after Annexin V/PI dual staining. Apoptotic rate was represented as a percentage of total cell populations. The proportion of dead cells (annexin V−/PI+), live cells (annexin V−/PI−), early apoptotic cells (annexin V+/PI−) and late apoptotic/necrotic cells (annexin V+/PI+) was measured for comparison; b histogram for statistical analysis shows the apoptosis rate of rabbit NP cells treated for 6, 12, 18, 24 and 36 h. The values are expressed as mean ± SD from three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001 vs. control, ANOVA/Dunnett’s T3 test); c representative fluorescence photomicrograph of Annexin V/PI dual staining output by confocal microscopy (Magnification ×200, scale bars represent 100 μm). The photographs of green and red fluorescence were taken under a same field and then were merged (Color figure online)
Compression promotes apoptotic morphological changes in NP cells
Under the inverted phase-contrast microscopy, apoptotic NP cells treated with compression after 36 h exhibited shedding of smaller fragments from the cells, cell shrinkage or threadlike morphology and almost detachment from the plates (Fig. 5a). As seen in Fig. 5b, the proportion of cells with brightly stained condensed nuclei slightly increased after compression treatment for 36 h as compared with the control group.

Morphologic changes in apoptotic rabbit NP cells. a Phase-contrast photomicrograph of rabbit NP cells stimulated with compression for 36 h. Apoptotic cells were characterized by shedding of smaller fragments from the cells, cell shrinkage or threadlike morphology and almost detachment from the plates (Magnification ×200, scale bars represent 100 μm); b after 36-h treatment of compression, apoptotic nuclei were condensed and brightly stained with Hoechst 33258 (Magnification ×200). Figures are representative of three independent experiments
Association of compression-induced apoptosis of NP cells with the inhibition of Bcl-2 and the activation of caspase-3 and Bax
Our results showed that the treatment of compression markedly increased Bax expression, but decreased Bcl-2 expression (Fig. 6a). In the assay of Bax gene, P value of group 6 h was 0.02, and less than 0.001 in other treatment groups. As to Bcl-2, all of the P values were less than 0.001. As shown in Fig. 6a, the expression of Bax mRNA began to increase at 6 h after treatment with compression and reached the maximum at 36 h among these groups. Western blot also revealed that the level of Bax protein was increased maximally after the same treatment for 36 h (Fig. 6b). We found that the mRNA expression of Bcl-2 was decreased gradually in the NP cells which were subjected to the treatment of compression. The reduction of Bcl-2 protein almost paralleled with that of Bcl-2 mRNA according to the analysis of Western blot (Fig. 6b). Furthermore, our results also indicated that the expression of caspase-3 at both protein and mRNA levels was obviously elevated in the NP cells which were subjected to compression (Fig. 6). The exact P values of each treatment group were 0.040, 0.005, 0.001, 0.008 and 0.002, respectively. These results implied that mitochondrial pathway was likely to be involved in the apoptotic process.

Effects of compression on the mRNA and protein expression of Bax, Bcl-2 and Caspase-3 in rabbit NP cells. a The mRNA levels of Bax, Bcl-2 and Caspase-3 detected by RT-PCR in rabbit NP cells subjected to compression at the magnitude of 1.0 MPa for different durations as mentioned above. The data of treated groups have been normalized to GAPDH and then shown as a ratio of normalized data to mRNA of control group. The values are expressed as mean ± SD from three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001 vs. control; ANOVA/LSD and Dunnett’s T3 test); b representative Western blot graphs for Bax, Bcl-2 and Caspase-3 in rabbit NP cells subjected to compression for 6, 12, 18, 24 and 36 h
The opening of MPTP triggered by compression
As demonstrated by many researches [28, 29], a significant feature of many apoptotic cascades is onset of the MPTP that results from an abrupt increase of permeability of the mitochondrial inner membrane to all molecules of less 1,500 Da. As shown in Fig. 7, the values of relative fluorescence intensity (RFI) detected by using fluorescence spectrophotometer were decreased in a time-dependent manner, demonstrating the increased activity of MPTP. And the P values of the five treatment groups were less than 0.001, suggesting there was significant difference between control group and each treatment groups.

The RFI of MPTP in different groups detected by fluorescence spectrophotometer. The reduction of RFI indicated the elevated activity of MPTP. The values are expressed as mean ± SD from three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001 vs. control, ANOVA/LSD test)
The increased ROS induced by compression
Mitochondria-generated ROS plays an important role in the release of cytochrome c and other pro-apoptotic proteins, which can trigger caspase activation and apoptosis. Excessive ROS can impair mitochondrial function and influence the viability of cells. As shown in Fig. 8, the level of ROS was enhanced gradually with the time prolonging. And all of the P values were less than 0.001. The result revealed that pressure treatment at different time lengths remarkably increased the level of ROS in NP cells.

Compression-induced increase of ROS level in a time-dependant pattern. The MFI of DCF was measured by flow cytometry and reflected the levels of intracellular ROS. Histogram for statistical analysis shows the ROS levels in rabbit NP cells treated for 6, 12, 18, 24 and 36 h. The data are expressed as mean ± SD of from three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001 vs. control, ANOVA/LSD test)
Decrease in MMP by compression
To further illustrate the involvement of mitochondria dysfunction in NP cells apoptosis, we measured MMP in the isolated rabbit NP cells by using flow cytometry and JC-1 staining in situ. The decline of MMP is considered as a symbolic event of early cellular apoptosis. The results from Fig. 9a and b showed that the treatment of compression reduced the MMP in a time-dependent manner, as indicated by a decrease in red (JC-1 aggregates)/green (JC-1 monomers) ratio. Moreover, P values of each treatment group were less than 0.001. As shown in Fig. 9c, the normal NP cells stained with JC-1 exhibited mitochondrial red fluorescence with a little green fluorescence, suggesting the cells in the normal polarization state. The JC-1 aggregates were dispersed to the monomeric form (green fluorescence) in the cells subjected to pressure at different time points as shown in Fig. 9c. The in MMP was decreased, as monitored by in situ JC-1 staining, primarily at the 24- and 36-h points after the treatment of pressure as compared with the normal group.

Compression-induced decrease in MMP of NP cells. a Representative dot plot of the changed MMP by flow cytometry after the labeling of fluorescent probe with JC-1. FL1-H Green, FL2-H Red; b the quantitative MMP from each group is expressed as the ratio of red fluorescence intensity over the green fluorescence intensity by flow cytometry. The data are expressed as mean ± SD from three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001 vs. control, ANOVA/Dunnett’s T3 test); c typical fluorescence photomicrograph of in situ JC-1 staining output by laser scan confocal microscopy (Magnification ×200, scale bars represent 100 μm) (Color figure online)
Discussion
A major cause of LBP is associated with the degeneration of IVD, of which the apoptosis of NP cells and the loss of ECM are the central features in the aged and degenerated IVD which is induced by multiple factors such as genetics, age and mechanical stimulus. In the present study we generally focus on the influence of mechanical loading on IVD cells as overload is supposed to accelerate the degeneration of IVD [11, 30]. A pressure of 1.0 MPa was widely adopted to induce IVD degeneration. Wang et al. [31] had once observed that static and dynamic compression may induce different biologic responses of the IVD. It has also been reported that the most significant cell apoptosis was noted in the discs which loaded at static 1.0 MPa. Wilke et al. [32] also reported that the mechanical pressure of human L4–L5 disc was 0.5 MPa for resting standing and 1.1 MPa for flexed-forward standing. Ariga et al. [33] found that the cartilage endplate cells apoptosis was induced using a static mechanical load (load weight, 1.0 MPa) in an organ culture system. In this study, we stated the question whether mitochondrial pathway is involved in the process of compression-induced apoptosis in NP cells. After treatment with compression, the decrease of proliferation and apoptosis of NP cells were induced. For the mitochondrial function, we found that the mitochondrial pathway may be involved in the process of compression-induced apoptosis of NP cells.
The biological characteristic of NP cells has been extensively analyzed for its central role in the early stage of IVD degeneration. So a large mount of culture systems of NP cells were used to simulate the degeneration of IVD. Many studies demonstrated that NP cells cultured in three-dimensional environment such as alginate system can maintain important characteristics of their phenotype, including the collagen and aggrecan biosynthesis and gene expression. However, the proliferation of NP cells was limited in this culture system. Furthermore, Wang et al. [34] found that gene expression levels of AF cells were altered in monolayer compared with alginate although the alteration was reversed when re-encapsulated in alginate, but NP cells appeared to be insensitive to the changes of culture conditions. At the same time, they also demonstrated that the feasibility of using the culture system in monolayer to increase the cell number. In addition, we also found that the NP cells before the third generation cultured in monolayer could maintain important characteristics of their phenotype, but the cells proliferated slowly and dedifferentiated when cultured in monolayer beyond the third generation. So we have chosen the cells of second generation in this study. To overcome this shortcoming, some investigators suggested that the NP cells which cultured in monolayer serial passages proliferated, and then re-encapsulating the NP cells in alginate would restore the characteristics of their phenotype. So it is necessary to perform further study by using this combined culture system in the later work.
Caspase-3 is a major executioner protease, responsible for initiating in the apoptotic program [35]. The caspase-3 is activated by pro-apoptotic molecules such as cytochrome c released from mitochondria, and the release of cytochrome c from mitochondria is inhibited by anti-apoptotic members of the Bcl-2 family of protein and stimulated by pro-apoptotic members such as Bax [17]. In our study, the expression of caspase-3 protein showed that the cleavage of procaspase-3 into the active form of caspase-3 occurred more obviously. A change in mitochondrial membrane permeability is essential for apoptosis, leading to translocation of apoptogenic cytochrome c and apoptosis-inducing factor into the cytoplasm. Such an event is regulated by the Bax and Bcl-2 proteins [36]. Furthermore, we found a markedly increased pro-apoptotic Bax protein with a concomitantly declined anti-apoptotic Bcl-2 protein. From these data, it could be possibly concluded that the compression induced apoptosis of NP cells by activating the mitochondrial pathway. More interestingly, we found the expression of Bax and Caspase-3 at both protein and mRNA levels and modest increase in apoptosis rate after the NP cells were subjected to compression for 12 h and beyond. Along with the extension of treatment, the alterations occurred in a time-dependent manner. But the increased level of caspase-3 gene expression was slightly lower in 6-h compression treatment group than other compression treatment groups. These findings revealed that the apoptosis of NP cells mediated by the mitochondrial pathway occurs as early as 12 h after the treatment of compression to NP cells.
Mitochondria-generated ROS plays a potent role in the release of cytochrome c and other pro-apoptotic proteins, which can trigger caspase activation and apoptosis [21, 37]. When the formation of ROS is enhanced, it can still influence the viability of cells and impair mitochondrial function, such as respiration and oxidative phosphorylation, mitochondrial permeability transition (MPT) and MMP. The proliferation, also the activity of mitochondrial dehydrogenase was inhibited by compression at this magnitude in a time-dependent pattern. The MPT remained something of biochemical curiosity until it was recognized in the late 1980s. Kroemer et al. [38] and coworkers proposed that the MPT is a critical event in the progression of apoptosis. The formation and opening of MPTP, which composed of adenine nucleotide translocator (ANT), cyclophilin-D (CYP-D) and voltage-dependent anion channel (VDAC), is considered the critical reason for the increase of permeability in mitochondria [39]. The decrease of MMP, the inhibition of respiration chain and the release of cytochrome c are the direct consequence of the opening in MPTP when cell damage is difficult to repair. Our results also revealed a decrease in the MMP, an increase in the amount of intracellular ROS and the opening of MPTP after treatment with the compression at each time point, which preceded the changes of mRNA and protein related to apoptosis. These findings suggest that the mitochondrial pathway is involved in apoptotic whole process of NP cells in response to treatment with compression.
In conclusion, our results revealed that the compression induces apoptosis in rabbit NP cells. Prolonged exposure of compression induced the apoptosis of NP cells, such as the increase in apoptosis rate, the activation of caspase-3 and Bax, and the inhibition of Bcl-2. In addition, the compression significantly suppressed the mitochondrial function, which was shown in the opening of MPTP, the excessive production of ROS, and the decreased MMP. These data demonstrate that the mitochondrial pathway participates in the apoptotic process of NP cells induced by compression. This study found the apoptosis of many NP cells, whereas impaired mitochondrial function of NP cells also was detected after compression, supporting that the mitochondrial pathway would be involved in the process of apoptosis induced by compression. These findings may have important implications on our understanding of the mechanism of NP cell apoptosis which contributes to the development of IVD degeneration. In addition, it will provide a new theoretical basis and ideas for improving the efficacy of gene therapy and tissue engineering treatment and delaying the recurrence of IVD degeneration.
References
Chen WH, Liu HY, Lo WC et al (2009) Intervertebral disc regeneration in an ex vivo culture system using mesenchymal stem cells and platelet-rich plasma. Biomaterials 30:5523–5533
Kalichman L, Hunter DJ (2008) The genetics of intervertebral disc degeneration. Familial predisposition and heritability estimation. Joint Bone Spine 75:383–387
Schultz DS, Rodriguez AG, Hansma PK et al (2009) Mechanical profiling of intervertebral discs. J Biomech 42:1154–1157
Zhao CQ, Wang LM, Jiang LS et al (2007) The cell biology of intervertebral disc aging and degeneration. Ageing Res Rev 6:247–261
Zhao CQ, Jiang LS, Dai LY (2006) Programmed cell death in intervertebral disc degeneration. Apoptosis 11:2079–2088
Setton LA, Chen J (2004) Cell mechanics and mechanobiology in the intervertebral disc. Spine 29:2710–2723
Aguiar D, Johnson SL, Osgema TR (1999) Notochordal cells interact with NP cells: regulation of proteoglycan synthesis. Exp Cell Res 246:129–137
Iwashina T, Mochida J, Miyazaki T et al (2006) Low-intensity pulsed ultrasound stimulates cell proliferation and proteoglycan production in rabbit intervertebral disc cells cultured in alginate. Biomaterials 27:354–361
Vonk LA, Kroeze RJ, Doulabi BZ et al (2010) Caprine articular, meniscus and intervertebral disc cartilage: an integral analysis of collagen network and chondrocytes. Matrix Biol 29:209–218
Hsieh AH, Twomey J (2010) Cellular mechanobiology of the intervertebral disc: new directions and approaches. J Biomech 43:137–145
Rannou F, Lee TS, Zhou RH et al (2004) Intervertebral disc degeneration: The role of the mitochondrial pathway in annulus fibrous cell apoptosis induced by overload. Am J Pathol 164:915–924
Zhao CQ, Liu D, Li H et al (2007) Interleukin-1β enhances the effect of serum deprivation on rat annular cell apoptosis. Apoptosis 12:2155–2166
Scott FL, Stec B, Pop C et al (2009) The Fas-FADD death domain complex structure unravels signaling by receptor clustering. Nature 457:1019–1022
Thornberry NA, Lazebnik Y (1998) Caspase: enemies within. Science 281:1312–1316
Wikstrom J, Twig G, Shirihai OS (2009) What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy. Int J Biochem Cell Biol 41:1914–1927
Lv L, Zhou ZX, Huang XZ et al (2010) Inhibition of peptidyl-prolyl cis/trans isomerase Pin1 induces cell cycle arrest and apoptosis in vascular smooth muscle cells. Apoptosis 15:41–54
Ott M, Gogvadze V, Orrenius S et al (2007) Mitochondria, oxidative stress and cell death. Apoptosis 12:913–922
Tripathy MK, Mitra D (2010) Differential modulation of mitochondria OXPHOS system during HIV-1 induced T-cell apoptosis: upregulation of Complex-IV subunit COX-II and its possible implications. Apoptosis 15:28–40
Haefen CV, Wendt J, Semini G et al (2011) Synthetic glycosidated phospholipids induce apoptosis through activation of FADD, caspase-8 and the mitochondrial death pathway. Apoptosis 16:636–651
Crawford N, Chacko AD, Savage KI et al (2011) Platinum resistant cancer cells conserve sensitivity to BH3 domains and obatoclax induced mitochondrial apoptosis. Apoptosis 16:311–320
Ravindran J, Gupta N, Agrawal M et al (2011) Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 16:145–161
Qian Y, Du YH, Tang YB et al (2011) ClC-3 chloride channel prevents apoptosis induced by hydrogen peroxide in basilar artery smooth muscle cells through mitochondria dependent pathway. Apoptosis 16:468–477
Kaneyama S, Nishida K, Takada T et al (2008) Fas ligand expression on human nucleus cells decreases with disc degeneration process. J Orthop Sci 13:130–135
Park JB, Lee JK, Park EY et al (2008) Fas/FasL interaction of NP and cancer cells with the activation of caspases. Int Orthop 32:835–840
Hutton WC, Elmer WA, Boden SD et al (1999) The effect of hydrostatic pressure on intervertebral disc metabolism. Spine 24:1507–1515
Zucchini-Pascal N, Sousa GD, Rahmani R (2009) Lindane and cell death: at the crossroads between apoptosis, necrosis and autophagy. Toxicology 256:32–41
Zhou YJ, Zhang SP, Liu CW et al (2009) The protection of selenium on ROS mediated-apoptosis by mitochondria dysfunction in cadmium-induced LLC-PK1 cells. Toxicol In Vitro 23:288–294
Rodriguez-Enriquez S, He L, Lemasters JJ (2004) Role of mitochondria permeability transition pores in mitochondria autophagy. Int J Biochem Cell Biol 36:2463–2472
Lemasters J, Nieminen AL, Qian T et al (1998) The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1366:177–196
Reza A, Nicoll SB (2008) Hydrostatic pressure differentially regulates outer and inner annulus fibrosus cell matrix production in 3D scaffolds. Ann Biomed Eng 36:204–213
Wang DL, Jiang SD, Dai LY (2007) Biologic response of the intervertebral disc to static and dynamic compression in vitro. Spine 32:2521–2528
Wilke HJ, Neef P, Caimi M et al (1999) New in vivo measurements of pressures in the intervertebral disc in daily life. Spine 24:755–762
Ariga K, Yonenobu K, Nakase T et al (2003) Mechanical stress-induced apoptosis of endplate chondrocytes in organ-cultured mouse intervertebral discs. Spine 28:1528–1533
Wang JY, Baer AE, Kraus VB et al (2001) Intervertebral disc cells exhibit differences in gene expression in alginate and monolayer culture. Spine 26:1747–1752
Li Z, Jo J, Jia JM et al (2010) Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141:859–871
Marzo I, Brenner C, Zamzami N et al (1998) Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281:2027–2031
Huang QR, Li Q, Chen YH et al (2010) Involvement of anion changer-2 in apoptosis of endothelial cells induced by high glucose through an mPTP-ROS-Caspase-3 dependent pathway. Apoptosis 15:693–704
Kroemer G, Pelit P, Zamzami N et al (1995) The biochemistry of programmed cell death. FASEB J 9:1277–1287
Crompton M (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem J 341:233–249
Acknowledgments
This work was supported by a grant from National Natural Sciences Foundation of China (No. 30700841, 81171149). Special thanks go to the members of the IVD degeneration research team at the Institute of Orthopedic Surgery of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, the People’s Republic of China for their help.
Conflict of interest
Foundation funds were received in support of this work. No benefits in any form have been or will be received from a commercial party related directly or indirectly to the subject of this manuscript. The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Fan Ding and Zeng-Wu Shao contributed equally to this work.
Rights and permissions
About this article
Cite this article
Ding, F., Shao, ZW., Yang, SH. et al. Role of mitochondrial pathway in compression-induced apoptosis of nucleus pulposus cells. Apoptosis 17, 579–590 (2012). https://doi.org/10.1007/s10495-012-0708-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-012-0708-3