
Abstract
Insulin-like growth factor 1 (IGF-1) inhibits 5-fluorouracil (5-Fu)-induced apoptosis in esophageal carcinoma cells; however, the mechanisms for IGF-1-induced 5-Fu chemoresistance remain unknown. In the human esophageal carcinoma cell line, CE48T/VGH, we show that IGF-1 up-regulated survivin expression at the post-transcriptional level and this up-regulation is mediated by both the PI3-K/Akt and casein kinase 2 signaling pathways. We then examine whether IGF-1-induced 5-Fu chemoresistance is mediated through up-regulation of survivin. Ectopic expression of survivin inhibits 5-Fu-induced apoptosis; furthermore, the abolition of survivin expression sensitizes cells to 5-Fu treatment and prevents the anti-apoptotic function of IGF-1 in esophageal carcinoma cell lines. We also found that ectopic expression of survivin or treatment with IGF-1 inhibits the release of Smac/DIABLO and caspases activation after 5-Fu treatment. Our results strongly suggest that IGF-1 inhibits 5-Fu induced apoptosis through increasing survivin levels, which prevents Smac/DIABLO release and blocks the activation of caspases. Therefore, up-regulation of IGF-1 and survivin would seem to be responsible for 5-Fu chemoresistance in esophageal cancer patients and these factors may be the valuable predictors of 5-Fu chemoresistance in esophageal carcinoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
5-Fluorouracil (5-Fu) is widely used in the treatment of a range of cancers, including esophageal carcinoma. However, the response rate for 5-Fu as a single treatment agent in esophageal carcinoma is only 15% and even for combinational chemotherapies or chemoradiation therapies, the response rate remains below 40% [1–3]. Thus, an understanding of the molecular mechanisms of resistance to 5-Fu is important for predicting or overcoming 5-Fu resistance in esophageal carcinoma. Various studies have identified several genes that contribute to 5-Fu resistance; these involve 5-Fu metabolism and the targets of 5-Fu, such as dihydropyrimidine dehydrogenase and thymidylate synthase [4]. Recently, the modulation of 5-Fu resistance by anti-apoptotic genes has gathered much attention. For example, down-regulation of the Bcl-2 family or the inhibitor of apoptosis protein (IAP) family sensitizes tumor cells to 5-Fu treatment [5–7].
Survivin, an IAP protein, has dual functions, namely regulating cell cycle progression and inhibiting apoptosis [8, 9]. The expression of survivin is undetectable in almost all adult normal tissues, however, survivin is over-expressed in various human tumors, including in esophageal carcinoma [10]. The presence of survivin in esophageal carcinoma is associated with an unfavorable patient prognosis and chemoresistance [11, 12]. However, the molecular mechanisms whereby survivin affects 5-Fu resistance have not been well investigated.
Unlike XIAP, which acts as direct inhibitor of caspases [13], survivin fails to bind caspases directly and the anti-apoptotic mechanisms of survivin rely on cooperation with cofactor molecules. For example, survivin can associate with HBXIP to suppress the activation of caspase-9 [14]. The survivin-XIAP interaction leads to stabilization of XIAP and subsequent enhancement of the anti-apoptotic function of XIAP [15]. In HeLa cells, survivin can interact with cytosolic Smac/DIABLO to prevent the formation of XIAP/Smac/DIABLO complex and further enhance the cytoprotection of XIAP [16]. Alternatively, survivin also can bind with mitochondrial Smac/DIABLO to delay its release from mitochondria and decrease the sensitivity of cancer cells to apoptosis stimulants [17].
In prostate cancer cells, IGF-1 up-regulates survivin through PI3-K/Akt pathway, thus mediating resistance to Flutamide [18]. The expression of IGF-1 receptor has been reported to be associated with an advanced tumor stage, a shorter survival and recurrence in esophageal squamous cell carcinoma [19]. In esophageal carcinoma cell lines, IGF-1 stimulation mediates 5-Fu chemoresistance [20]. However, the molecular mechanisms underlying this effect have not been well defined.
In this study, we show that through the PI3-K/Akt and casein kinase 2 (CK2) signaling pathways, IGF-1 modulates survivin protein stability and that survivin inhibits 5-Fu-induced apoptosis by preventing Smac/DIABLO release from mitochondria, which sequentially blocks caspase activation. More importantly, survivin is essential to protecting cells from 5-Fu-induced apoptosis by IGF-1.
Results
IGF-1 up-regulates survivin
IGF-1 is the survival factor for several chemotherapy drugs in esophageal carcinoma cells [20]. Here we found, in CE48T/VGH cells, that IGF-1 decreased serum starvation and 5-Fu induced apoptosis (Fig. 1a). To identify the molecules participating in the IGF-1 preventing 5-Fu-induced apoptosis, we first examined the protein levels of several pro- and anti-apoptotic molecules after treatment with IGF-1. As shown in Fig. 1b, 5-Fu treatment decreased the protein level of survivin by 86%; however, IGF-1 increased the survivin protein level and reversed the effect of 5-Fu. In contrast to this change, treatment with IGF-1 or 5-Fu did not obviously affect the cIAP, Bcl-2 and Bax protein levels.

IGF-1 inhibits 5-Fu induced apoptosis and up-regulates the expression of survivin in CE48T/VGH cells. (a) Cells were incubated in serum-free medium (SF) containing 400 μM 5-Fu without or with 100 ng/ml IGF-1 and then apoptosis was analyzed. (b) Cells were treated as described in (a) and Western blot analysis was performed at 6 h after treatment. * p < 0.05, unpaired t-test
IGF-1 up-regulates survivin at the post-transcriptional level in CE48T/VGH cells
To assess the mechanisms by which IGF-1 regulates survivin expression, we first measured the survivin mRNA and protein levels after IGF-1 treatment over time. Upon IGF-1 treatment, the level of survivin protein was found to be significantly increased at 3 h and this was sustained for 12 h in CE48T/VGH cells; however, there were no visible effects on cIAP, Bcl-2 and Bax (Fig. 2a). In contrast, there was no obvious change in the level of survivin mRNA after IGF-1 treatment for up to 24 h (Fig. 2b). To further investigate the impact of IGF-I on survivin protein level, cycloheximide was used to block de novo protein synthesis. In the presence of cycloheximide, the survivin protein level was reduced 60% at 6 h; while upon IGF-1 stimulation, there was only a slight decrease (13%) in the survivin protein level (Fig. 2c). When taken together, these results indicated that IGF-1 up-regulated survivin expression at the post-transcriptional level.

IGF-1 up-regulates survivin at the post-transcriptional level. CE48T/VGH cells were treated with IGF-1 for the indicated time intervals and (a) protein expressions were analyzed by Western blotting; (b) survivin mRNA was analyzed by real-time PCR. (c) Four hours after treatment with IGF-1, cells were incubated with 50 μg/ml cycloheximide (CHX), harvested at the indicated time intervals and survivin protein was analyzed by Western blotting (left panel). The protein expression levels of survivin were quantified (right panel)
IGF-1 regulates survivin expression via the PI3-K/Akt and CK2 signaling pathways
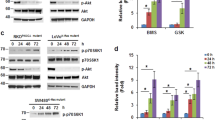
We then investigated the signaling pathways through which IGF-1 exerted the up-regulation of survivin protein expression. Two inhibitors of PI3-K, LY294002 and Wortmannin, were found to significantly inhibit the IGF-1’s effect on the up-regulation of survivin protein (Fig. 3a). Akt is a common down stream substrate for PI3-K during anti-apoptosis signaling. To analyze the role of Akt in this regulation, adenoviral vectors containing an Akt dominant negative mutant (Ad-Akt-DN) were used. In the Ad-Akt-DN-infected cells, but not in the non-infected or the adenoviral vector infected cells, IGF-1-up-regulation of survivin protein expression was inhibited (Fig. 3b). We also found that IGF-1 treatment significantly activated CK2 kinase activity in parallel to the increase in survivin protein level (Fig. 3c). The effect of IGF-1 on up-regulating survivin protein expression was abolished by CK2 kinase inhibitors, tetrabromobenzotriazole and quinalizarin (1,2,5,8-tetrahydroxyanthraquinone) [21, 22], in a dose dependent manner (Fig. 3c, d). In contrast, treatment with the ERK inhibitor, PD98059, had no effect on the IGF-1 induced up-regulation of survivin protein expression (Fig. 3e). The above findings indicated that the PI3-K/Akt and CK2 signal pathways were critical to the IGF-1 induced up-regulation of survivin expression.

IGF-1 regulates survivin through the PI3-K/Akt and CK2 pathways. CE48T/VGH cells were pretreated with (a) LY294002 (LY; 20 μM) or Wortmannin (Wor; 100 nM); (c) tetrabromobenzotriazole (TBB); (d) quinalizarin (e) PD98059 (PD; 25 μM) for 2 h before adding IGF-1 to the culture medium. The cell lysates were analyzed using Western blotting and the kinase activity of CK2 was determined in (c). (b) Non-infected cells (Non) and cells infected with the adenoviral vector (Adv) or Ad-Akt-DN were treated with IGF-1. Cell lysates were analyzed using Western blotting
Survivin plays an important role in the anti-apoptotic function of IGF-1 in esophageal carcinoma cells
To investigate whether survivin is a mediator associated with IGF-1 preventing 5-Fu-induced apoptosis, we first evaluated if survivin is able to inhibit 5-Fu-induced apoptosis. Ectopic expression of survivin, using the adenoviral vector containing the survivin gene (Ad-survivin), resulted in the modified cells showing resistance to 5-Fu; the elevation in CE48T/VGH cells was from a LD50 of 0.395 mM to a LD50 of 0.787 mM (Fig. 4a). In addition, when survivin was ectopic expressed in CE48T/VGH cells, serum-starvation and 5-Fu induced apoptosis was significantly decreased compared to the control (Fig. 4b). In agreement with these findings, the protein levels of the activated forms of caspase 3, 8 and 9 were increased after 5-Fu treatment in the CE48T/VGH cells but not in the survivin ectopic expressed cells (Fig. 4c). These results showed that survivin enhanced 5-Fu resistance in CE48T/VGH cells.

Survivin plays an important role in the anti-apoptotic function of IGF-1 in CE48T/VGH cells. After infected with indicated adenoviruses, cells were treated with 5-Fu and (a) cell viabilities; (b) apoptosis; (c) processions of caspases were assayed. (d) After infection with indicated adenoviruses, cells were incubated in SF containing 5-Fu without or with IGF-1 and then apoptosis was analyzed. * p < 0.05; NS not significant difference; unpaired t-test
Next, we tested if survivin is essential for the IGF-1 anti-apoptotic function. We abolished survivin expression by infected cells with the adenoviral vector containing the antisense-survivin gene (Ad-anti-survivin) and then determined the anti-apoptotic effect of IGF-1. In the survivin knockdown cells, 5-Fu-induced apoptosis was significantly increased (Fig. 4d, columns 3 and 7) and IGF-1 failed to prevent 5-Fu-induced apoptosis (Fig. 4d, columns 7 and 8).
To ensure we were not looking a phenomenon specific to CE48T/VGH, we tested whether survivin is also required for IGF-1 to prevent 5-Fu-induced apoptosis in other esophageal carcinoma cells. A 5-Fu sensitive cell line, CE81T/VGH, and a 5-Fu resistant cell line, TE-2, were used. In these two cell lines, IGF-1 increased the survivin protein level and reversed the down-regulation effect of 5-Fu on survivin (Fig. 5a). Survivin overexpression in TE-2 and CE81T/VGH cells, significantly conferred cells resistant to 5-Fu treatment; the LD50 was elevated from 0.085 to 0.136 mM and from 0.779 to 1.364 mM in CE81T/VGH and TE-2 cells, respectively (Fig. 5b, c). In accordance with results in CE48T/VGH cells, the presence of IGF-1 inhibited 5-Fu-induced apoptosis in sh-luc(control)-infected CE81T/VGH and TE-2 cells (Fig. 5d, e, columns 3 and 4). Down-regulation of survivin by sh-survivin in CE81T/VGH and TE-2 cells (to 40% and 60%, respectively, Fig. 5d, e), 5-Fu-induced apoptosis was significantly increased (Fig. 5d, e, columns 3 and 7) and IGF-1 only partially inhibited 5-Fu-induced apoptosis (Fig. 5d, e, columns 7 and 8). Taken together, these results clearly showed that survivin is able to protect esophageal carcinoma cells from 5-Fu-induced apoptosis and survivin is an important mediator in the anti-apoptotic function of IGF-1 in esophageal carcinoma cells.

Survivin plays an important role in the anti-apoptotic function of IGF-1 in CE81T/VGH and TE-2 cells. (a) Cells were incubated in SF medium containing 5-Fu without or with IGF-1 for 6 h and then Western blot analysis was performed. (b, c) After infected with indicated adenoviruses, cells were treated with 5-Fu and cell viabilities were assayed. (d, e) Indicated cells were incubated in SF containing 5-Fu without or with IGF-1 and then apoptosis was analyzed. * p < 0.05; unpaired t-test
Survivin inhibits 5-Fu-induced apoptosis by preventing the release of Smac/DIABLO from the mitochondria
Then, we sought to explore the role of Smac/DIABLO in survivin inhibition of 5-Fu-induced apoptosis. As shown in Fig. 6a, 5-Fu treatment significantly increased the release of Smac/DIABLO and cytochrome c from the mitochondria in a time-dependent manner. On the ectopic expression of survivin in CE48T/VGH cells, survivin was localized to both the cytoplasm and the mitochondria and, after 5-Fu exposure, there was complete inhibition of the release of Smac/DIABLO from the mitochondria; however, the release of cytochrome c was not affected (Fig. 6b). These results indicated that survivin suppressed the release of Smac/DIABLO from mitochondria and this sequentially inhibited 5-Fu-induced apoptosis.

Survivin inhibits 5-Fu-induced apoptosis by preventing the release of Smac/DIABLO from the mitochondria in CE48T/VGH cells. The subcellular fractions were prepared for Western blot analysis after (a) cells were treated with 5-Fu for the indicated time intervals; (b) cells were treated with 5-Fu for 48 h after indicated adenovirus infection; (c) cells were incubated in SF containing 5-Fu without or with IGF-1
As IGF-1 induced an up-regulation of survivin, we sought to analyze if IGF-1 treatment has the same effect on Smac/DIABLO as survivin. Figure 6c shows that IGF-1 treatment blocked the release of Smac/DIABLO that is induced by 5-Fu. Moreover, IGF-1 treatment also inhibited the 5-Fu-induced release of cytochrome c from mitochondria.
Discussion
In summary, this study explores one molecular mechanism of IGF-1 chemoresistance via the up-regulation of survivin expression and shows that survivin inhibits 5-Fu-induced apoptosis by preventing Smac/DIABLO release from the mitochondria, which then blocks apoptosis. Survivin is abundantly expressed in virtually every human tumor that has been tested, but is expressed at low to undetectable levels in most normal tissues. The deregulation of survivin expression in cancers is involved in transcriptional and post-transcriptional control [23]. As lowering of survivin expression enhances the cytotoxic effects of chemotherapy drugs, much attention has been devoted towards elucidating pathways controlling survivin level in tumor cells [23]. In this study, we found that, in esophageal carcinoma cells, IGF-1 up-regulates survivin expression through the PI3-K/Akt and CK2 signaling pathways. This regulation is remarkable because it pinpoints a new survivin anti-apoptotic network that is initiated by IGF-1/CK2 signaling, and it depends on stabilizing survivin protein but not on gene transcription as previous reports. In prostate cancer cells, IGF-1/Akt/mTOR signaling increases the stability and translation of survivin mRNA; in HEK 293T cells, CK2 increases survivin expression via β-catenin–Tcf/Lef-mediated transcription [24, 25]. These data, along with ours, indicate that IGF-1 mediates diverse regulation mechanisms to influence survivin expression in different cell types.
Phosphorylation is an important post-translational regulatory mechanism to affect protein stability and cytoprotective functions of survivin [26, 27]. Since there are two putative CK2 phosphorylation sites, Thr48 and Thr97, on survivin [28], we initially speculate that IGF-1 may regulate survivin protein stability through CK2 phosphorylation. Unfortunately, both Akt and CK2 failed to phosphorylate survivin in vitro (data not shown), which suggests that Akt and CK2 might regulate other molecules that control survivin stability. Possibilities include Hsp60, Hsp90 or aryl hydrocarbon receptor interacting protein (AIP), which have been reported to bind to survivin and maintain survivin stability against proteasome-dependent degradation [29–31]. In our data, we also observed that LY294002, Wortmannin and PD98059 treatment increased the levels of survivin protein for 40%, 60% and 40%, respectively in CE48T/VGH cells. Since survivin is a radio- and chemo-resistance factor, several chemicals have been reported to induce survivin protein expression due to stabilization of the protein, i.e. etoposide [17], adriamycin, paclitaxel [32, 33] and cisplatin [17, 34], suggesting that this change in survivin expression could be a possible general effect of these drugs. However, the mechanisms for the increase in survivin level caused by LY294002, Wortmannin and PD98059 need further investigation.
Autocrine stimulation by IGF-1 is involved in mediating multiple chemotherapy drugs resistance in esophageal carcinoma cells [20], while the molecular mechanisms for IGF-1 induced chemoresistance is not well investigated. This is the first study to extend the important role of survivin in IGF-1 mediated 5-Fu chemoresistance in esophageal carcinoma cells. First, the over-expression of survivin significantly reduced the cytotoxic effect of 5-Fu. Second, inhibition of survivin expression not only increased the cytotoxic effect of 5-Fu but also abrogated IGF-1 mediated 5-Fu resistance, which indicates that survivin is an important mediator in IGF-1-induced 5-Fu resistance. Taken together, up-regulation of survivin is one of the mechanisms by which IGF-1 mediates 5-Fu resistance.
The cytoprotective effect of survivin relies on translocation to the mitochondria and intermolecular cooperation [14, 15, 27]. In this study, ectopic expression of survivin increases the amount of survivin in mitochondria. This specifically blocked the release of Smac/DIABLO but not cytochrome c from the mitochondria. Although 5-Fu-induced cytochrome c release is not affected, ectopic expression of survivin still inhibits 5-Fu-induced caspase activation. These data indicate that the anti-apoptotic mechanisms of survivin connect to two parallel pathways: (1) mitochondrial survivin may associate with Smac/DIABLO in a way that prevents its release from mitochondria and sequesters it away from other IAP proteins, such as XIAP, and further remove the pro-apoptotic function of Smac/DIABLO [16, 17], (2) cytosolic survivin may suppress cytochrome c-induced apoptosis by interacting with HBXIP or XIAP in a manner that prevents the caspase 9 activation during 5-Fu induced apoptosis [14, 15] (Fig. 7).

Schematic illustrating the role of survivin in IGF-1 mediating 5-Fu resistance in esophageal carcinoma cells. IGF-1 goes through PI3 K/Akt and CK2 signaling pathways to up-regulate survivin and prevent 5-Fu-induced Smac/DIABLO release and caspase activations
Our results indicate that the increased levels of survivin mediated by the IGF-1 counteract the anti-tumor action of 5-Fu. Thus, 5-Fu may not have a particular benefit in tumors in which the survivin levels are known to be elevated. Our finding implies that survivin may be therapeutically valuable as well as being a predictive biomarker of 5-Fu chemoresistance in esophageal carcinoma. Consequently, combined therapies, including suppression of survivin expression by, for example, YM155 and a blockade of IGF-1R signaling by, for instance a monoclonal antibody against IGF-1R, CP-751,871, may be promising therapeutic approaches that target esophageal carcinoma cells [35, 36].
Materials and methods
Reagents
Recombinant human IGF-1 was obtained from Intergen (Purchase, NY, USA). 5-fluorouracil, PD98059, LY294002, Wortmannin, cycloheximide and digitonin were purchased from Sigma–Aldrich (St Louis, MO, USA). Tetrabromobenzotriazole and quinalizarin (1,2,5,8-tetrahydroxyanthraquinone) were bought from Calbiochem (La Jolla, CA, USA). Antibodies against G3PDH and caspase 9 were purchased from Upstate Biotechnology Inc. (Lake Placid, NY, USA). Antibodies against Bcl-2, Bax, phospho-Akt (Ser473), Akt, phospho-Erk, Erk, Smac/DIABLO and caspase 8 were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibody against cIAP was bought from AnaSpec Inc. (San Jose, CA, USA). Antibody against CK2 was obtained from Santa Cruz (Santa Cruz, CA, USA). Antibody against caspase 3 was purchased from Stressgen Bioreagents Co. (Ann Arbor, MI, USA). Antibodies against Cytochrome c and Heat shock protein 60 were bought from R&D Systems (Minneapolis, MN, USA).
Cell lines, RNA preparation and real-time PCR analysis
The human esophageal carcinoma cell line, CE48T/VGH and CE81T/VGH were established in our laboratory [37] and routinely cultured in Dulbecco’s modified eagle’s medium (DMEM) with 5% fetal calf serum. TE-2 was kindly provided by Dr Morio Kasai, The Second Department of Surgery, Tohoku University School of Medicine, Sendai, Japan and routinely cultured in DMEM with 10% fetal calf serum.
Total RNA from CE48T/VGH cells was purified and cDNA was reversed transcribed from 1 μg total RNA as described previously [38]. Real-time PCR reactions using SYBR GREEN PCR Master Mix (Applied Biosystems, Foster City, CA, USA) were run on an ABI Prism 7000 PCR machine. The primers used in this study were as follows; survivin forward: 5′-TCAGCCCAACCTTCA CATCTG-3′; survivin reverse: 5′-CTGCTGCCTCCAAAGAAAGC-3′. DDX-5 forward: 5′-GAATTTCACTGAACCCACTGCTATT-3′; DDX-5 reverse: 5′-TGCCACTCCAACCATATCCA-3′.
Adenovirus and lentivirus constructions
Replication-deficient adenoviruses encoding GFP (Ad-IE), survivin (Ad-survivin) or antisense survivin (Ad-anti-survivin) were generated using the Adeno-X Expression System I (Clontech Laboratories, Mountain View, CA, USA). The survivin cDNA was amplified from Jurket total RNA and cloned into pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA). The survivin DNA fragment was then cloned into pShuttle IE/R vector and further cloned into pAdeno-X vector.
The packaging system for lentiviral production included the vectors, pCMV-∆R8.91, pMD.G, and the shRNA expression plasmid (pLKO.1), were obtained from the National RNAi Core Facility, Taiwan. For the knockdown of survivin, the target sequence 5′-CCGCATCTCTACATTCAAGA-3′ (TRCN0000073718) was inserted into pLKO.1 vector. A shRNA vector against luciferase (pLKO.1-shLuc) was used as a negative control for knockdown validation. To generate lentiviral particles, a mixture of the 3 vectors, pCMV-∆R8.91, pMD.G, and pLKO.1, was prepared and transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The virus suspension was collected at 48 and 36 h after transfection. For lentivirus infection, culture medium was replaced with medium containing 8 μg/ml polybrene before lentivirus infection. After 24 h infection, 2 μg/ml puromycin was added for 3 days to select successful infection cells.
Western blotting
The same amount of protein was subjected to 10% or 15% SDS–PAGE and the resolved proteins were transferred onto 0.22 μM PVDF membranes (Millipore, Billerica, MA, USA). Antigen–antibody complexes were visualized with peroxidase-conjugated secondary antibodies using the ECL system (Amersham Pharmacia, Bucks, UK). The polyclonal anti-survivin antibodies were generated from immunized rabbits using recombinant survivin protein in our laboratory.
Subcellular fractionation
The cytosolic and mitochondrial fractions were generated using a digitonin-based subcellular fractionation technique [39, 40]. Briefly, cells were harvested by trypsin treatment and then digitonin-permeabilized for 10 min on ice in cytosolic extraction buffer (250 mM sucrose, 70 mM Tris pH 7.0, 20 mM PMSF, 1 μg/ml leupeptin, 2 μg/ml aprotinin, 100 μg/ml digitonin). After centrifuged at 800 g for 10 min at 4°C, the supernatant (cytosolic fraction) was collected and the pellet was dissolved for 10 min on ice in mitochondrial lysis buffer (50 mM HEPES, 4 mM EDTA, 2 mM EGTA, 1% Triton X-100, 20 mM PMSF, 1 μg/ml leupeptin, 2 μg/ml aprotinin). After centrifugated at 2,300 g for 10 min at 4°C, the supernatant comprising the mitochondria-enriched fraction was retained.
Viability assay
Cells were seeded into each well of 24-well plates overnight and infected with indicated viruses in 2% FCS-medium for overnight, and then the cells were incubated in SF medium for 24 h. Subsequently, the indicated drugs were added to the cells for another 48 h and the viable cells were counted by the trypan blue exclusion.
Apoptosis assay
Apoptosis was analyzed using the Cell Death Detection ELISA (Roche Diagnostics, Mannheim, Germany) to measure cytoplasmic histone-associated DNA fragments following the manufacturer’s instruction. Briefly, cells were seeded into each well of 96-well plates. After the indicated treatments, both the floating and attached cells were lysed in lysis buffer and incubated for 30 min at room temperature. The supernatant was then transferred into an ELISA microtiter plate for analysis.
CK2 activity assay
The CK2 kinase activity was measured using the CK2 Assay Kit (Upstate Biotechnology Inc.), according to the supplier’s instructions. Briefly, cells were collected and lysed on ice for 30 min in NET buffer (50 mM Tris pH 7.4, 1 mM EDTA, 150 mM NaCl, 0.5% NP-40, 20 mM PMSF, 2 μg/ml aprotinin, 1 μM leupeptin, 1 mM sodium vanadate, 80 mM sodium pyrophosphate, and 50 mM sodium fluoride). Whole cell homogenates were centrifuged at 15,500 g for 30 min at 4°C and the supernatants were collected. Next, 1 mg of the proteins was reacted with anti-CK2α antibody coated protein G/A beads (Calbiochem) overnight at 4°C. The immunoprecipitated samples were dissolved in 20 μl of NET buffer and 10 μl was subjected to SDS–PAGE to measure the efficiency of the immunoprecipitation; the other 10 μl was incubated with substrate peptide in assay dilution buffer I containing 10 μCi [γ-32P]-ATP for 10 min at 30°C. The reaction was then stopped by adding 40% trichloroacetic acid solution. Aliquots were then transferred onto P81 paper squares and the radioactivity was measured using a scintillation counter.
References
Enzinger PC, Ilson DH, Kelsen DP (1999) Chemotherapy in esophageal cancer. Semin Oncol 26:12–20
Sumpter K, Harper-Wynne C, Cunningham D et al (2005) Report of two protocol planned interim analyses in a randomised multicentre phase III study comparing capecitabine with fluorouracil and oxaliplatin with cisplatin in patients with advanced oesophagogastric cancer receiving ECF. Br J Cancer 92:1976–1983
Akutsu Y, Matsubara H, Shuto K et al (2009) Clinical and pathologic evaluation of the effectiveness of neoadjuvant chemoradiation therapy in advanced esophageal cancer patients. World J Surg 33:1002–1009
Longley DB, Harkin DP, Johnston PG (2003) 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3:330–338
Graidist P, Phongdara A, Fujise K (2004) Antiapoptotic protein partners fortilin and MCL1 independently protect cells from 5-fluorouracil-induced cytotoxicity. J Biol Chem 279:40868–40875
Zhu H, Guo W, Zhang L et al (2005) Bcl-XL small interfering RNA suppresses the proliferation of 5-fluorouracil-resistant human colon cancer cells. Mol Cancer Ther 4:451–456
Zhang S, Ding F, Luo A et al (2007) XIAP is highly expressed in esophageal cancer and its downregulation by RNAi sensitizes esophageal carcinoma cell lines to chemotherapeutics. Cancer Biol Ther 6:973–980
Ambrosini G, Adida C, Sirugo G, Altieri DC (1998) Induction of apoptosis and inhibition of cell proliferation by survivin gene targeting. J Biol Chem 273:11177–11182
Li F, Ambrosini G, Chu EY et al (1998) Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 396:580–584
Altieri DC (2003) Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 22:8581–8589
Rosato A, Pivetta M, Parenti A et al (2006) Survivin in esophageal cancer: an accurate prognostic marker for squamous cell carcinoma but not adenocarcinoma. Int J Cancer 119:1717–1722
Kato J, Kuwabara Y, Mitani M et al (2001) Expression of survivin in esophageal cancer: correlation with the prognosis and response to chemotherapy. Int J Cancer 95:92–95
Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC (1999) Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J 18:5242–5251
Marusawa H, Matsuzawa S, Welsh K et al (2003) HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J 22:2729–2740
Dohi T, Okada K, Xia F et al (2004) An IAP-IAP complex inhibits apoptosis. J Biol Chem 279:34087–34090
Song Z, Yao X, Wu M (2003) Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J Biol Chem 278:23130–23140
Ceballos-Cancino G, Espinosa M, Maldonado V, Melendez-Zajgla J (2007) Regulation of mitochondrial Smac/DIABLO-selective release by survivin. Oncogene 26:7569–7575
Zhang M, Latham DE, Delaney MA, Chakravarti A (2005) Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene 24:2474–2482
Imsumran A, Adachi Y, Yamamoto H et al (2006) Insulin-like growth factor-I receptor as a marker for prognosis and a therapeutic target in human esophageal squamous cell carcinoma. Carcinogenesis 28:947–956
Liu YC, Leu CM, Wong FH et al (2002) Autocrine stimulation by insulin-like growth factor I is involved in the growth, tumorigenicity and chemoresistance of human esophageal carcinoma cells. J Biomed Sci 9:665–674
Cozza G, Mazzorana M, Papinutto E et al (2009) Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase CK2. Biochem J 421:387–395
Sarno S, Reddy H, Meggio F et al (2001) Selectivity of 4, 5, 6, 7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (‘casein kinase-2’). FEBS Lett 496:44–48
Altieri DC (2008) Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer 8:61–70
Vaira V, Lee CW, Goel HL, Bosari S, Languino LR, Altieri DC (2006) Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene 26:2678–2684
Tapia JC, Torres VA, Rodriguez DA, Leyton L, Quest AF (2006) Casein kinase 2 (CK2) increases survivin expression via enhanced {beta}-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc Natl Acad Sci USA 103:15079–15084
O’Connor DS, Grossman D, Plescia J et al (2000) Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci USA 97:13103–13107
Dohi T, Xia F, Altieri DC (2007) Compartmentalized phosphorylation of IAP by protein kinase A regulates cytoprotection. Mol Cell 27:17–28
Altieri DC, Marchisio PC (1999) Survivin apoptosis: an interloper between cell death and cell proliferation in cancer. Lab Invest 79:1327–1333
Kang BH, Altieri DC (2006) Regulation of survivin stability by the aryl hydrocarbon receptor-interacting protein (AIP). J Biol Chem 281:24721–24727
Fortugno P, Beltrami E, Plescia J et al (2003) Regulation of survivin function by Hsp90. Proc Natl Acad Sci USA 100:13791–13796
Ghosh JC, Dohi T, Kang BH, Altieri DC (2008) Hsp60 regulation of tumor cell apoptosis. J Biol Chem 283:5188–5194
Ling X, Bernacki RJ, Brattain MG, Li F (2004) Induction of survivin expression by taxol (paclitaxel) is an early event, which is independent of taxol-mediated G2/M arrest. J Biol Chem 279:15196–15203
Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC (2003) Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res 63:230–235
Brenes O, Arce F, Gatjens-Boniche O, Diaz C (2007) Characterization of cell death events induced by anti-neoplastic drugs cisplatin, paclitaxel and 5-fluorouracil on human hepatoma cell lines: possible mechanisms of cell resistance. Biomed Pharmacother 61:347–355
Iwasa T, Okamoto I, Suzuki M et al (2008) Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in non-small cell lung cancer cell lines. Clin Cancer Res 14:6496–6504
Cohen BD, Baker DA, Soderstrom C et al (2005) Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751, 871. Clin Cancer Res 11:2063–2073
Hu CP, Hsieh HG, Chien KY et al (1984) Biologic properties of three newly established human esophageal carcinoma cell lines. J Natl Cancer Inst 72:577–583
Wong FH, Hu CP, Chiu JH et al (1994) Expression of multiple oncogenes in human esophageal carcinomas. Cancer Invest 12:121–131
Waterhouse NJ, Goldstein JC, von AO, Schuler M, Newmeyer DD, Green DR (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol 153:319–328
Ekert PG, Silke J, Hawkins CJ, Verhagen AM, Vaux DL (2001) DIABLO promotes apoptosis by removing MIHA/XIAP from processed caspase 9. J Cell Biol 152:483–490
Acknowledgments
This work was supported in part by grants from the National Science Council (NSC93-3112-B-010-026; NSC94-3112-B-010-012; NSC95-3112-B-010-003) and Ministry of Education, Aim for the Top University Plan (95A-C-T04-56; 96A-D-T137; 98A-C-D127) to F-H. Wong. We are grateful to Dr. Chen-Kung Chou (Graduate Institute of Basic Medical Science, Chang-Gung University, Taiwan) for helpful comments on the manuscript. We thank Dr. Michael Hsiao (Genomic Research Center of Academia Sinica, Taiwan) for the Ad-Akt-DN vector and the VGH National Yang-Ming University Genome Research Center (YMGC) for pOTB7-Smac plasmid.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Juan, HC., Tsai, HT., Chang, PH. et al. Insulin-like growth factor 1 mediates 5-fluorouracil chemoresistance in esophageal carcinoma cells through increasing survivin stability. Apoptosis 16, 174–183 (2011). https://doi.org/10.1007/s10495-010-0555-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-010-0555-z