
Abstract
The structure and function of the mitochondrial network is regulated by mitochondrial biogenesis, fission, fusion, transport and degradation. A well-maintained balance of these processes (mitochondrial dynamics) is essential for neuronal signaling, plasticity and transmitter release. Core proteins of the mitochondrial dynamics machinery play important roles in the regulation of apoptosis, and mutations or abnormal expression of these factors are associated with inherited and age-dependent neurodegenerative disorders. In Parkinson’s disease (PD), oxidative stress and mitochondrial dysfunction underlie the development of neuropathology. The recessive Parkinsonism-linked genes PTEN-induced kinase 1 (PINK1) and Parkin maintain mitochondrial integrity by regulating diverse aspects of mitochondrial function, including membrane potential, calcium homeostasis, cristae structure, respiratory activity, and mtDNA integrity. In addition, Parkin is crucial for autophagy-dependent clearance of dysfunctional mitochondria. In the absence of PINK1 or Parkin, cells often develop fragmented mitochondria. Whereas excessive fission may cause apoptosis, coordinated induction of fission and autophagy is believed to facilitate the removal of damaged mitochondria through mitophagy, and has been observed in some types of cells. Compensatory mechanisms may also occur in mice lacking PINK1 that, in contrast to cells and Drosophila, have only mild mitochondrial dysfunction and lack dopaminergic neuron loss. A better understanding of the relationship between the specific changes in mitochondrial dynamics/turnover and cell death will be instrumental to identify potentially neuroprotective pathways steering PINK1-deficient cells towards survival. Such pathways may be manipulated in the future by specific drugs to treat PD and perhaps other neurodegenerative disorders characterized by abnormal mitochondrial function and dynamics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Mitochondrial structure and function
Among the most important functions of mitochondria are the synthesis of adenosine triphosphate (ATP) through oxidative phosphorylation, fatty acid oxidation, regulation of redox and calcium signaling, and the control of apoptosis in response to a variety of adverse extracellular and intracellular signals [1]. As descendants of ancient prokaryotes, mitochondria have inner and outer membranes that separate the matrix from the intermembrane space (IMS) and the IMS from the cytosol. The inner mitochondrial membrane (IMM) is highly folded into the cristae and harbors the respiratory chain complexes of the electron transport chain and ATP synthase. Cells contain many mitochondria, each carrying several copies of a small circular mitochondrial genome (16,569 bp in humans) in the matrix. The mammalian mitochondrial genome contains 37 genes, which encode 13 protein subunits of the respiratory complexes I, III, IV and V (ATP synthase) as well as 22 tRNAs and 2 rRNAs that are required for the translation of mitochondrial transcripts. The remainder of the 900–1000 estimated mitochondrial proteins [2], as well as additional tRNAs required for translation, are nucleus-encoded and must be imported into mitochondria via an organelle-specific import machinery [3].
Mitochondrial fission and fusion
Mitochondria can change their shape, size and inner membrane structure in a dynamic fashion. In addition, the mitochondria of a single cell do not function in isolation, but form a complex reticulum whose morphology undergoes continuous changes in response to metabolic stimuli and signaling pathways. These changes are regulated by fission and fusion events that remodel mitochondrial membranes. Several excellent reviews have been published recently on the machinery and regulation of mitochondrial dynamics [4–11]. Therefore, I will limit the discussion on the core proteins and most important regulatory mechanisms involved in these processes.
The molecular machinery of mitochondrial fission and fusion is best characterized in yeast [4]. In mammalian cells, mitochondrial fission depends on the GTPase dynamin-related protein (Drp1) [12–14]. To promote fission, Drp1 must translocate from the cytosol to the outer mitochondrial membrane (OMM), where it oligomerizes into ring-like structures and constricts the mitochondria through GTP hydrolysis. In addition, fission requires OMM proteins including hFis1 and others [5, 11, 15–21]. Control of mitochondrial fission occurs primarily through post-translational modifications of Drp1, as well as direct Drp1-interacting proteins that increase the mitochondrial pool of Drp1 [10, 22]. In proliferating cells mitochondria must undergo fission during mitosis, which is promoted by Cdk1/cyclin B-dependent phosphorylation of Drp1 on Ser-585 (rat Drp1; Ser-618 in human Drp1) [23]. In addition, the conserved residue Ser-637 (human Drp1; Ser-656 in rat Drp1) is phosphorylated by cyclic AMP-dependent protein kinase (PKA) and dephosphorylated by calcineurin [24]. Expression of the phosphomimetic Ser656Asp Drp1 mutant resulted in a marked elongation and perinuclear clustering of mitochondria, while non-phosphorylatable Ser656Ala Drp1 caused mitochondrial fragmentation [24, 25]. In agreement with this, dephosphorylation of Drp1 by calcineurin enhanced the translocation of Drp1 to mitochondria and promoted fission [26]. Thus, the phosphorylation status of Ser-637 (Ser-656 in rats) determines Drp1 localization and activity [24, 26–28]. In addition, both sumoylation [29–32] and ubiquitination [33] affect fission. Drp1 interacts with Ubc9 and Small ubiquitin-like modifier 1 (Sumo1), and transient transfection of YFP-tagged Sumo1 into cells dramatically increased mitochondrial fission, with a portion of YFP-Sumo1 colocalizing with endogenous Drp1 at mitochondrial fission sites [29]. In contrast, overexpression of SUMO1/sentrin- specific peptidase 5 (SENP5) rescued Sumo1-induced mitochondrial fission, partly through destabilization of Drp1, while silencing of SENP5 expression increased mitochondrial fission [31]. Recently, mitochondrial-anchored protein ligase (MAPL) has been identified as the first mitochondrial SUMO E3 ligase [32]. Overexpression of MAPL stimulates fission [34], and MAPL binds to Drp1 in vivo and sumoylates Drp1 in vitro [32]. Conversely, silencing of MAPL leads to a decrease in the levels of Drp1 and SUMO conjugates [32]. These experiments show that MAPL-dependent sumoylation of Drp1 is associated with increased fission [29, 31, 32]. The ubiquitin E3 ligase MITOL/MARCH-V is an integral protein of the OMM with its RING finger domain exposed to the cytosol. MITOL has been shown to ubiquitinate both hFis1 and Drp1, increasing their turnover [35]. Conversely, depletion of MITOL by RNAi resulted in increased mitochondrial fragmentation, and MITOL overexpression could rescue mitochondrial fragmentation induced by overexpression of hFis1, suggesting that MITOL1 is an inhibitor of fission [35]. However, the results of another group published more recently pointed in the opposite direction, because cells with silenced MARCH-V expression showed abnormally elongated mitochondria with increased interconnectivity, indicative of reduced fission and/or increased fusion. The latter group proposed that wildtype MARCH-V facilitates the assembly and/or disassembly of functional fission complexes on the OMM through ubiquitination of Drp1, thereby promoting fission [36]. Finally, MARCH-V was also shown to bind to Mfn2 and to promote mitochondrial fusion in an Mfn2-dependent manner [37]. Thus, while MITOL/MARCH-V clearly affects mitochondrial morphology, further studies are necessary to clarify its exact mechanism.
Mitochondrial fusion relies on the coordinated action of three GTPases. Mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) are integral outer membrane proteins that tether organelles for OMM fusion by mediating homotypic and heterotypic interactions between adjacent mitochondria [38–40]. Inner membrane fusion is mediated by optical atrophy 1 (Opa1), which is an IMM protein with a soluble IMS form generated through proteolytic processing [40–42]. In addition, Opa1 plays an important role in the maintenance and regulation of IMM structure [41, 43, 44]. In yeast, several proteins with regulatory functions in mitochondrial fusion have been identified [10]. For example, the IMS protein Ups1 regulates the alternative topogenesis and sorting of Mgm1 (the yeast homologue of Opa1) in the IMM under certain metabolic conditions [45]. PRELI (protein of relevant evolutionary and lymphoid interest) is the human orthologue of Ups1 [46], but whether it fulfills a similar function in mammals is unknown.
Importance of mitochondrial transport and subcellular distribution for neuronal function
In neurons, mitochondria are particularly abundant at synapses and in unmyelinated axon segments of the peripheral nervous system [47–50]. Mitochondria concentrate and are recruited to subcellular regions with high metabolic requirements, for example in the vicinity of active growth cones in neurons [51]. Pharmacological and genetic studies have demonstrated that mitochondria regulate neuronal activity, synaptic plasticity and neurotransmitter release by controlling synaptic Ca2+ concentrations and ATP-dependent vesicle movements [47, 52–59]. In addition, manipulation of Drp1 and Opa1 function that reduced dendritic mitochondrial content in hippocampal neurons caused the loss of synapses and dendritic spines, demonstrating the importance of subcellular mitochondrial positioning for the development and morphological plasticity of neuronal processes [51]. Trafficking of mitochondria in neurons relies on the molecular motor proteins kinesin-1 (KIF1B) and kinesin-3 (KIF5B) as well as cytosolic dynein [10, 49, 60]. In Drosophila, the two proteins Miro and Milton function as an adaptor complex that anchors mitochondria to kinesin-1 [53, 58, 61]. Miro is a GTPase anchored to the outer mitochondrial membrane, whereas Milton is a cytosolic protein that binds to Miro and kinesin heavy chain [61]. Mammalian Miro recruits Grif-1 (the mammalian homologue of Milton) to mitochondria in a GTPase-dependent way, resulting in enhanced transport of mitochondria towards the distal ends of hippocampal processes [62]. Miro also plays an important role as a calcium sensor and the regulation of calcium-dependent mitochondrial transport [63, 64]. The interaction of Miro with kinesin is calcium-sensitive, and increased Ca2+ concentrations in the physiological range disrupt the interaction of Miro with KIF5, resulting in mitochondrial arrest [64]. In addition, upon activation of N-methyl-D-aspartate (NMDA) receptors Miro positioned mitochondria at the postsynaptic site, where calcium concentrations and energy demand were high [64]. Drosophila dMiro mutants retained their mitochondria in the neuron soma and failed to distribute the organelles to axons, whereas dMiro overexpression resulted in the accumulation of mitochondria in terminal boutons of the fly neuromuscular junction [53]. In mammalian cells, overexpression of Miro increased mitochondrial trafficking in both anterograde and retrograde direction by increasing the overall proportion of mobile mitochondria [65]. In contrast, RNAi-mediated Miro gene silencing decreased the number of mobile mitochondria [62]. Collectively, these experiments show that Miro and Milton regulate mitochondrial positioning to sites of increased energy usage in a calcium-dependent manner [66, 67]. Other recently identified proteins affecting mitochondrial transport are syntabulin, which connects mitochondria to KIF5B and is involved in activity-dependent anterograde transport of mitochondria to presynaptic sites [68, 69] and syntaphilin, which serves as a docking receptor for mitochondria in axons and may allow an increased concentration of mitochondria to be maintained in the synapse [50]. Finally, both ectopic expression and knockdown of the E3 ubiquitin ligase MULAN interfered with mitochondrial trafficking and morphology, suggesting that mitochondrial transport is also regulated by ubiquitin-dependent processes [70].
Regulation of apoptosis by mitochondrial fission and fusion proteins
Apoptosis is accompanied by changes in mitochondrial morphology. Often, mitochondrial fission is an early event during apoptosis and it has been shown that silencing of hFis results in increased net fusion and resistance to staurosporine-, actinomycin D- or anti-Fas-induced apoptosis [71]. Likewise, inhibiting mitochondrial fission by expression of dominant-negative Drp1K38A decreased apoptosis, albeit to a lesser degree [71]. Silencing of hFis1 expression blocked apoptosis by inhibiting the translocation of cytosolic Bax to mitochondria [71], while Drp1K38A reduced cytochrome c release and apoptosis without significantly blocking Bax translocation [71, 72]. However, although mitochondrial fission often coincides with apoptosis, fission per se is dispensible for apoptosis [73, 74]. In addition, fission and the release of cytochrome c and other pro-apoptotic molecules can occur independently from each other. First, cells depleted of both hFis1 and Opa1 were resistant to apoptosis despite showing extensive mitochondrial fission [71]. Second, a recently developed chemical Drp1 inhibitor (mdivi-1) was shown to block mitochondrial outer membrane permeabilization (MOMP) induced by cleaved Bid in a cell-free in vitro assay with purified mitochondria, in which mitochondrial fission was unlikely to occur [75]. Mdivi-1 prevents the assembly of Drp1 complexes on the outer membrane of mitochondria, suggesting that Drp1 may be important for the induction of MOMP independent of mitochondrial fission [75, 76]. Third, although the anti-apoptotic proteins Bcl-xL and Mcl-1 could inhibit the release of cytochrome c in cells overexpressing Bax/Bak, they did not reverse the mitochondrial fragmentation, indicating that MOMP and mitochondrial fission are separable events [77, 78]. It has been shown that Bax and Bak colocalize with mitochondrial fission sites, Drp1 and Mfn2 during apoptosis [79]. These Bax/Bak-dependent apoptotic fission complexes are very stable and coincide with Drp1 sumoylation [80]. Taken together, these data implicate Drp1 in the regulation of apoptosis through influencing MOMP. MOMP may be accompanied or enhanced by mitochondrial fission, but does not strictly depend on it [74] (Fig. 1).

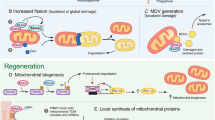
Mitochondrial fission and fusion proteins in apoptosis. Mitochondrial outer membrane permeabilization (MOMP) is stimulated by Bax/Bak-dependent formation of Sumo-modified Drp1 complexes at the OMM. In healthy cells Drp1 shuttles between mitochondrial fission sites and the cytosol. At the beginning of apoptosis, Drp1 gets locked to the OMM and forms stable, Bax/Bak-dependent complexes that promote MOMP. In addition, Bax and Bak become colocalized with Drp1 complexes as well as Mfn2 punctae on the OMM. Bax-dependent sumoylation of Drp1 coincides with apoptosis and may be required for Drp1 complex stabilization, suggesting that Sumo modification of Drp1 may enhance apoptosis. However, Drp1 sumoylation also stimulates fission, which can occur during apoptosis but is not a prerequisite for cell death. Genetic manipulations (Drp1 RNAi) or chemical inhibitors (mdivi-1) that reduce Drp1 complex formation or stability decrease MOMP. Silencing of hFis1 expression and expression of a dominant-active form of the fusion protein Mfn2 inhibit translocation of Bax to mitochondria and Bax activation, while Opa1 RNAi has the opposite effect. Therefore, manipulations that increase net fusion reduce the release of cytochrome c and apoptosis, while fission proteins can increase apoptosis. In normal cells, an Opa1 complex at the IMM maintains the cristae junctions impermeable for cytochrome c, thereby preventing most of the cytochrome c molecules from reaching the OMM, where they can be released into the cytosol after MOMP. During apoptosis, the Opa1 complex becomes disassembled in a Bax/Bak-dependent manner, which results in a structural change of the cristae junctions and the release of the cristae cytochrome c pool into the remainder of the IMS, with access to OMM “pores”. Opa1 complex disassembly may also occur upon dissipation of Δψm and proteolytic processing, which correlates with conversion of longer to shorter Opa1 isoforms. In addition, excess mitochondrial calcium may promote Opa1 complex disassembly. For details and references, please see main text
In contrast, mitochondrial fusion proteins conferred protection against apoptosis [71, 72, 81, 82]. For example, knocking down Opa1 expression enhanced sensitivity to apoptosis, characterized by Bax translocation to mitochondria, release of cytochrome c, nuclear fragmentation and caspase activation [71]. Furthermore, a dominant-active mutant of Mfn2 with reduced GTP hydrolysis activity promoted increased fusion and interfered with the translocation and activation of Bax, thereby protecting mitochondria against free radical-induced permeability transition [72]. Finally, several studies showed that Opa1, through its role in maintaining cristae structure and junctions, attenuates the release of cytochrome c from mitochondria [81, 83, 84]. Cristae are tubular structures that trap about 85% of the cytochrome c molecules within the folds of the IMS, with their “neck” or junction stabilized by a complex of Opa1 oligomers (Fig. 1). Frezza et al. showed that induction of apoptosis by Bid correlated with the disruption of Opa1 oligomers that keep the cristae junctions impermeable for cytochrome c, indicating remodeling of an Opa1 complex at the cristae junction during apoptosis [85]. Yamaguchi et al. confirmed this result, although in this study a narrowing of the cristae junctions was observed after Opa1 complex disassembly [86]. In addition, MOMP occurred under these conditions, as expected [86]. Opa1 complex disassembly and MOMP resulted in the release of cytochrome c, Smac/DIABLO and Opa1, which could be inhibited by small molecule drugs against serine proteases of the OMM [86]. Interestingly, two of these drugs were able to block MOMP but failed to inhibit the disassembly of Opa1 complexes [86]. In addition, overexpression of Opa1Q297V, a mutant that is resistant to disassembly, inhibited cristae junction remodeling, cytochrome c release and apoptosis in transiently transfected 293T cells, without blocking activation of Bax and induction of MOMP [86]. Collectively, these results show that Bax/Bak-induced disassembly of Opa1 complexes and MOMP can occur independently from each other, and that both events are necessary for the release of the complete mitochondrial pool of cytochrome c and Opa1 [84, 86] (Fig. 1). Although cristae junction remodeling and MOMP can occur independently, it has been hypothesized that activated Bax/Bak act through some OMM proteins that transmit the signal to Opa1 at the IMM, either directly or indirectly via a protein in IMS [86]. In an earlier study it was shown that Bik-induced cristae remodeling could be inhibited by expression of dominant-negative Drp1K38E [87], indicating that Drp1 may relay the death signal to Opa1 in the IMM. In addition, inhibition of mitochondrial calcium uptake also interfered with Bik-induced opening of cristae [87]. In thapsigargin-treated cells undergoing ER stress, Opa1 and cytochrome c release required mitochondrial permeability transition (MPT) and Bax, as well as the matrix peptidyl-prolyl-isomerase cyclophilin-D [88]. However, because calcium-induced MPT also occurred with purified mitochondria stripped of their outer membrane and the voltage-dependent anion channel, it was proposed to be an inner membrane phenomenon directly affecting Opa1 release [88].
Additional regulation of Opa1 function can occur through post-transcriptional and post-translational mechanisms. The Opa1 primary transcript undergoes alternative splicing [89] and at least five Opa1 protein isoforms have been detected in mammalian cells [90]. Based on exon-specific RNAi experiments, it has been suggested that Opa1 transcripts containing exon 4 are important for mitochondrial fusion, while mRNAs containing exons 4b or 5b encode Opa1 isoforms that regulate cytochrome c release [91]. The IMM protease presenelin-associated rhomboid-like (PARL) is involved in the generation of a soluble Opa1 isoform present in the IMS [92]. Mitochondria from PARL-deficient mice display faster cristae remodeling and cytochrome c release during apoptosis, showing that Opa1 processing influences apoptosis [92, 93]. Consistent with this idea, it has recently been shown that deletion of the prohibitin-2 (Phb2) gene in mice, which leads to the selective loss of long isoforms of Opa1 and aberrant cristae morphogenesis, renders cells more vulnerable to apoptosis [94, 95]. However, prohibitins most likely act as scaffolding proteins at the IMM and are not proteases themselves, and their effect on Opa1 processing is thus indirect. Finally, the expression of the mitochondrial AAA protease paraplegin and dissipation of Δψm resulted in the proteolytic conversion of long to short Opa1 isoforms and mitochondrial fragmentation [96, 97]. However, RNAi experiments in HeLa cells showed that only Yme1, another m-AAA protease, is required for the constitutive processing of a subset of longer Opa1 isoforms and that Yme1 depletion results in increased fusion and mitochondrial interconnectivity [98].
In summary, Opa1 plays a critical role in the regulation of the structure and tightness of cristae junctions, where Opa1 complex disassembly at the IMM causes the release of cytochrome c from cristae. Cristae junction remodeling may be further regulated through constitutive and induced processing of Opa1 by mitochondrial proteases and Δψm. Several other proteins affecting mitochondrial dynamics have been implicated in apoptosis regulation. These factors, and a more detailed overview of the relationship of mitochondrial structure and apoptosis have been described recently in reviews specifically dedicated to this topic [11, 99].
Mitochondrial function and dynamics in Parkinson’s disease
Rapidly growing evidence implicates impaired mitochondrial function and dynamics in the pathogenesis of PD. Data supporting a causal relationship between abnormal mitochondrial function and dynamics, cell death and dopaminergic neuron degeneration has come from a host of studies that addressed the functions of Parkin, PINK1 and other proteins linked to recessive familial Parkinsonism in cultured cells, flies and mice [100–102]. Collectively, these experiments have shown that Parkin and PINK1 not only influence the function and morphology of mitochondria, but also promote the degradation of dysfunctional organelles and possibly regulate the transport of mitochondria in axons. In the following sections, I will discuss the activities of Parkin and PINK1 relevant to these processes, and describe metabolic and anatomic features of the dopaminergic system that may explain its exquisite vulnerability to perturbations in mitochondrial function and dynamics.
Parkin maintains mitochondrial integrity and protects against oxidative and unfolded protein stress
Mutations in the Parkin (PARK2) gene are associated with autosomal recessive juvenile Parkinsonism (AR-JP) [103, 104]. The E3 ubiquitin protein ligase Parkin plays a role in the proteasome-mediated turnover of several proteins in vitro [105–108], some of which accumulate in the brains of AR-JP patients and/or mice carrying a targeted deletion of the Parkin gene, showing that they are authentic Parkin substrates in vivo [109–111]. Drosophila lacking Parkin display increased sensitivity to oxidative stress, dopaminergic neuron loss, and severe structural mitochondrial abnormalities in muscle and germline tissues associated with apoptotic muscle degeneration and male sterility [112–114]. Ablation of Parkin in mice resulted in respiratory defects, reduced expression of subunits of respiratory complexes I and IV, decreased levels of anti-oxidant proteins, increased protein and lipid peroxidation [115], and abnormalities in dopamine neurotransmission, but no loss of dopaminergic neurons [116, 117]. Parkin-deficient zebrafish showed selective dopamine neuron loss associated with complex I impairment [118]. Fibroblasts from patients with Parkin mutations showed reduced mitochondrial connectivity, complex I activity and ATP synthesis, and were more sensitive to rotenone-induced death [119]. In agreement with these findings, Parkin overexpression increased mitochondrial membrane potential, selectively enhanced the expression of mitochondrial complex I subunits and reduced accumulation of reactive oxygen species (ROS) in mitochondria of transfected cells [120]. Interestingly, transgenic flies overexpressing familial PD-linked mutants of Parkin (R275 W and Q311X) also exhibited dopaminergic neurodegeneration, vacuolization of mitochondria, concentric membraneous structures within mitochondria and disruption of cristae [121, 122]. Likewise, age-dependent loss of nigral dopaminergic neurons and striatal dopamine fibers were observed in BAC-transgenic mice expressing ParkinQ311X, along with accumulation of proteinase K-resistant α-synuclein and progressive hypokinesia [123]. These studies suggest that certain Parkin mutations may cause dopamine neurodegeneration by a toxic gain-of-function.
Recently, Parkin has been linked to DNA repair [124, 125]. Parkin-deficient fibroblasts exhibited reduced DNA excision repair in assays where a luciferase reporter construct, which had been mutagenized in vitro by UV light or incubation with H2O2, was transfected into wildtype or Parkin-/- fibroblasts [124]. The defect in DNA excision repair could be restored by cotransfection of wildtype, but not pathogenic mutant Parkin, into Parkin-/- cells [124]. As a possible mechanism, it was shown that Parkin interacts with the proliferating cell nuclear antigen (PCNA), which coordinates DNA excision repair, and that inhibiting this interaction with a dominant-negative Parkin fragment also reduced the ability of Parkin to stimulate DNA repair [124]. A recent study with SH-SY5Y neuroblastoma cells showed that overexpression of Parkin enhanced replication and transcription of mitochondrial DNA, and protected mitochondrial DNA against oxidative damage by stimulating mitochondrial DNA repair [125].
Taken together, these data implicate Parkin in several diverse neuroprotective pathways, including ubiquitin-dependent degradation of toxic proteins, inhibition of oxidative stress, maintenance of mitochondrial structure, function and DNA integrity, as well as the stimulation of presynaptic dopamine neurotransmission.
PINK1 acts as a kinase and protects against oxidative stress
Homozygous mutations in the PARK6 gene, that encodes the protein PTEN-induced kinase 1 (PINK1), are the second most frequent cause for autosomal recessive early-onset Parkinsonism (EOPD) [126–131]. Recently, certain compound heterozygous PINK1 mutations have been found in patients with late-onset sporadic PD [132]. PINK1 is a ubiquitously expressed serine/threonine kinase with a short N-terminal mitochondrial targeting sequence that directs import of PINK1 into mitochondria [126, 133–137]. However, PINK1 has also been detected in the cytosol [133, 134, 138, 139], and one group showed substantial amounts of endogenous PINK1 in the microsome-rich fraction co-localized with calnexin, suggesting an association with the endoplasmatic reticulum [139]. The formation of the cytosolic PINK1 isoforms depends on import and processing of full-length PINK1 within mitochondria [140]. Within mitochondria, PINK1 has been localized both to the IMM facing the IMS [133, 140], and the OMM with the kinase domain facing the cytosol [141]. It has been shown that EOPD-associated mutations at least partially reduce PINK1 kinase activity, either by directly affecting the kinase domain or destabilizing PINK1 [134, 142, 143]. However, familial PD mutations do not interfere with mitochondrial import of PINK1 [133, 134]. Three PINK1 substrates have been identified to date. These are the mitochondrial protease HtrA2 [144], the mitochondrial chaperone TRAP1 (Hsp75) [142] and Parkin [145]. Both HtrA2 and TRAP1 are present in the IMS, while most of Parkin is located in the cytosol. HtrA2 is phosphorylated by PINK1 on serine 142 in response to the activation of the p38 stress-signaling pathway, and phospho-mimetic HtrA2 mutants suggest that this phosphorylation increases HtrA2 protease activity [144]. Increased HtrA2 protease activity may mediate neuronal survival under stress, as suggested by the phenotype of HtrA2 knockout mice, which develop neurodegeneration in the striatum, leading to a disorder with a Parkinsonian phenotype and death at 1 month of age [146]. Additionally, the observation that phosphorylation of serine 142 of HtrA2 is significantly reduced in the brains of PD patients with PINK1 mutations shows that HtrA2 is a true PINK1 substrate in vivo [144]. Phosphorylation of TRAP1 by PINK1 is necessary to protect cultured cells against oxidative stress-induced cytochrome c release and apoptosis [142], and overexpression of TRAP1 in the rat brain reduced oxidative stress and infarct size after focal ischemia and improved mitochondrial function under these conditions [147]. Finally, in cells PINK1 directly phosphorylates Parkin on threonine 175, which causes a striking redistribution of Parkin from the cytosol to mitochondria [145]. As discussed later, PINK1 and Parkin function in a common pathway to maintain the integrity of the mitochondrial network, by preventing mitochondrial dysfunction (PINK1, Parkin) and promoting degradation of defective mitochondria through autophagy (Parkin).
Effects of PINK1 ablation on mitochondrial function
Several groups have generated mammalian cells with stable or transient PINK1 gene silencing by RNA interference (RNAi). Characterization of these cells revealed reduced mitochondrial membrane potential [148–151], increased oxidative stress [149, 151–153], increased sensitivity to Parkinsonian toxins and complex I inhibitors [150, 154–156], reduced respiration and ATP synthesis [153, 157, 158], decreased mitochondrial DNA levels and respiration [149] and proteasomal deficits associated with α-synuclein aggregation [158]. Mitochondrial defects were also detected in fibroblasts and immortalized lymphoblasts from PD patients with G309D and W437Stop PINK1 mutations, which displayed lower respiratory activity, decreased activity of cytochrome c oxidase and elevated levels of lipid peroxidation [159, 160]. Recently, it has been shown that mitochondria of PINK1-deficient neurons accumulate higher basal levels of Ca2+ in the matrix (due to reduced calcium efflux capacity) and display reduced mitochondrial Ca2+ storage capacity associated with Ca2+ overload [151]. As a consequence, PINK1-deficient neurons were significantly more vulnerable to Ca2+-induced loss of Δψm and subsequent permeability transition pore opening [151]. In addition, increased production of ROS was observed secondary to mitochondrial Ca2+ overload [151].
Effects of PINK1 ablation on mitochondrial morphology
The consequences of PINK1 gene silencing on mitochondrial morphology and dynamics have been controversial and apparently contradictory. However, several groups have observed inner membrane abnormalities, such as decreased cristae density and disorganized or swollen cristae [148–150, 152]. Enlarged and swollen mitochondria were detected in human neurons after PINK1 gene silencing by RNAi [149]. In COS7 cells, which possess dynamic mitochondrial networks overexpression of PINK1 increased the number of cells with fragmented mitochondria [161], whereas simultaneous overexpression of PINK1 and dominant-negative Drp1K38A resulted in more cells with long tubular mitochondria [161]. Likewise, several lines of PINK1-deficient Drosophila developed prominent mitochondrial abnormalities, such as fragmented cristae and hollow-appearing mitochondria, swollen and enlarged mitochondria, as well as dopamine neuron death and apoptotic muscle degeneration [162–164]. Overexpression of the mitochondrial fission protein Drp1 rescued mitochondrial abnormalities and other defects in flies, while heterozygous loss-of-function mutations in the drp1 gene were mostly lethal in the absence of PINK1 or Parkin. In contrast, reduced expression of the mitochondrial fusion proteins Opa1 and Mfn2 significantly suppressed muscle degeneration and abnormal mitochondrial morphology in the absence of PINK1 and Parkin [161, 164, 165]. These results show that, in Drosophila and certain mammalian cells, shifting the net mitochondrial dynamics towards fission is protective, while decreased fission has the opposite effect. This has led to the suggestion that PINK1 promotes fission and/or inhibits fusion [165].
However, inconsistent with this interpretation, several groups have reported increased mitochondrial fission in cells lacking PINK1 [148, 150, 152]. For example, human M17 neuroblastoma cells with a lentiviral vector-mediated stable PINK1 knockdown exhibited mitochondrial fragmentation, which was exaggerated by expression of Drp1 and rescued by Drp1 RNAi, suggesting that PINK1 antagonizes mitochondrial fission and/or promotes fusion [150]. The same authors also showed that the amount of Ser-637 phosphorylated Drp1 is reduced by about 30% in PINK1-deficient cells due to the activation of the calcium-dependent phosphatase calcineurin [150]. Moreover, the calcineurin inhibitor FK506 blocked both Drp1 dephosphorylation and mitochondrial fragmentation [150]. Dephosphorylation of Drp1 by calcineurin was shown by others to enhance the translocation of Drp1 to mitochondria and promote fission [26]. However, despite increased calcineurin activity and reduced Drp1 phosphorylation, the mitochondrial levels of Drp1 were not higher in M17 cells with silenced PINK1 expression compared to control cells [150]. This suggests that mitochondrial fragmentation in these cells is not a consequence of increased mitochondrial Drp1 recruitment. However, silencing of PINK1 was associated with increased GTPase activity of Drp1 in vitro [150], which may increase fission in vivo. Another group recently showed that silencing of PINK1 by siRNAs resulted in a higher percentage of cells containing truncated and fragmented mitochondria [157]. These alterations in mitochondrial morphology were associated with reduced ATP synthesis and could be reversed by overexpression of Mfn2, Opa1 or dominant-negative Drp1K38E [157]. Collectively, these experiments show that lack of PINK1 in cultured cells results in mitochondrial fragmentation that depends on the fission protein Drp1 and can be ameliorated by overexpression of fusion proteins Mfn2 and Opa1 [157].
In mice, deletion of the PINK1 gene resulted in reduced evoked dopamine release and impaired corticostriatal long-term potentiation and long-term depression, in the absence of dopaminergic neuron degeneration [166]. Furthermore, mice devoid of PINK1 exhibit selective mitochondrial defects in the striatum, with an increased number of large mitochondria, impaired activities of respiratory complexes I and II, and reduced aconitase activity [167]. In addition, an age-dependent functional decay of cortical mitochondria was observed [167]. Respiration of partially purified cortical mitochondria was reduced more significantly by oxidative stress and heat shock in the absence of PINK1 [167], suggesting an increased sensitivity of PINK1-deficient mitochondria to exogenous stressors. Mitochondria from mice expressing low levels of abnormal PINK1 transcripts as a result of the attempt to target the G309D mutation to the endogenous PINK1 gene exhibited progressive reduction in mitochondrial preprotein import and reduced respiration, as well as increased aggregation of mitochondria under proteasomal stress [168]. Primary neurons from PINK-/- mice, and human dopaminergic neurons with silenced PINK1 expression develop widespread mitochondrial dysfunction, such as reduced Δψm and morphological abnormalities, increased oxygen radical production and heightened vulnerability to apoptosis [149]. Interestingly, human neurons with silenced PINK1 expression showed compensatory adaptations, including increased numbers of mitochondria, age-dependent elevated expression of specific respiratory complexes, and increased total citrate synthase activity [149].
Relationship of mitochondrial function and dynamics and pathways affected in PINK1-deficient cells
Because mitochondrial function and dynamics influence each other, it is difficult to study whether mitochondrial dysfunction in PINK1-deficient cells is a cause or consequence of abnormal mitochondrial morphology. However, the discrepant findings with respect to mitochondrial morphology in cells and animals lacking PINK1, as discussed above, suggest that alterations in mitochondrial morphology may occur secondary to mitochondrial dysfunction.
Several groups observed a reduced Δψm in cells devoid of PINK1 (see above). Changes in Δψm play important roles in regulating size and axonal transport of mitochondria [169], and chemical uncouplers that disrupt the Δψm block mitochondrial transport [170]. In cultured embryonic chicken dorsal root ganglion neurons, 90% of mitochondria with high Δψm were transported towards the growth cone, whereas 80% of mitochondria with low Δψm were transported towards the cell body, demonstrating that the Δψm determines the direction of mitochondrial transport in these neurons [170]. While FK506 rescued mitochondrial fragmentation in M17 human neuroblastoma with silenced PINK1 expression (presumably by inhibition of calcineurin-mediated dephosphorylation of Drp1), it only partially ameliorated the deficit in Δψm, supporting the view that the alterations in mitochondrial connectivity occurred secondary to the impaired Δψm [150].
Abnormal cristae structure is another defect frequently observed in PINK1-deficient cells (see above). PINK1 was recently shown to interact with mitofilin [171], a mitochondrial IMS protein that acts as an organizer of cristae morphology and junctions [172]. This suggests the possibility that PINK1 controls cristae structure via mitofilin. In the absence of PINK1, abnormal cristae morphology and junctions may enhance the vulnerability of cells to apoptosis.
Dysregulated mitochondrial calcium homeostasis may also contribute to changes in mitochondrial dynamics. Calcium efflux from mitochondria is compromised in PINK1-deficient mammalian neurons due to a defect of the mitochondrial Na+/Ca2+ exchanger [151]. This results in mitochondrial calcium overload and in turn increased ROS production via activation of NADPH oxidase, inhibition of the glucose transporter and impaired respiration [151]. Moreover, mitochondria from PINK1-deficient neurons were more susceptible to calcium-induced opening of the mitochondrial permeability transition pore [151]. Interestingly, increased mitochondrial fission caused by overexpression of Drp1 protected against calcium-induced mitochondrial abnormalities and subsequent apoptosis [173]. This raises the possibility that mitochondrial fragmentation in PINK1−/− cells may be a protective, adaptive response to mitochondrial calcium overload.
Lastly, oxidative stress could affect mitochondrial dynamics in PINK1-deficient cells. Mitochondrial fragmentation and upregulation of autophagy in PINK1−/− cells were mediated by mitochondrial oxidant production [152]. Mitochondria from the cerebral cortex of PINK1−/− mice were more susceptible to H2O2-induced respiratory defects, suggesting reduced ROS resistance of PINK1-deficient mitochondria [167]. It has recently been proposed that PINK1 may have a critical function in the oxidative stress defense downstream of PI3 kinase/Akt signaling and activation of FOXO transcription factors [174], which play important roles in protecting cells against oxidative stress via upregulation of several antioxidant enzymes [175]. FOXO3a binds to the PINK1 promoter and activates transcription of the PINK1 gene [174]. Transcriptional activation of PINK1 by FOXO3a attenuates the loss of reduced glutathione (GSH) in cytokine-deprived lymphocytes and increases lymphocyte survival [174]. Consistent with these results, GSH levels and mitochondrial respiration were reduced in cells after PINK1 gene silencing [153], and the amount of oxidized glutathione (GSSG) was significantly elevated in fibroblasts from PD patients harboring the familial G309D PINK1 mutation [159].
In summary, a major role of PINK1 is to maintain mitochondrial integrity by preventing calcium- and oxidative stress-induced mitochondrial dysfunction, loss of Δψm, and abnormal cristae structure (Fig. 2). In the absence of PINK1, impaired mitochondrial function may lead to secondary changes in mitochondrial dynamics, such as mitochondrial fragmentation, which may require increased Drp1 activity. While excessive fission likely exaggerates mitochondrial defects [150], limited fission can have protective effects as seen in Drosophila [161, 164, 165]. This may be attributed to the stimulatory effects of fission on mitophagy [176], respiration and ATP synthesis [177, 178], maintenance of mitochondrial DNA [178] and the transport of mitochondria to synapses during synaptogenesis [179]. It is not clear whether fusion may have been activated in those PINK1-deficient cell lines with swollen and enlarged mitochondria [149] and in Drosophila lacking PINK1. Fusion inhibits mtDNA abnormalities [180, 181] and is neuroprotective [182]. However, PINK1-deficient flies developed dopaminergic neuron loss and muscle degeneration, suggesting that the enlarged mitochondrial phenotype may have been caused by a mechanism other than enhanced fusion.

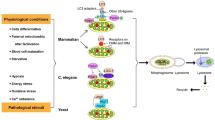
Early mitochondrial defects and postulated downstream effects and compensatory mechanisms in cells devoid of PINK1. Early defects likely include impaired Δψm, abnormal cristae morphology and calcium overload, resulting in increased mitochondrial ROS production, decreased respiration and enhanced sensitivity to apoptosis due to facilitated membrane permeability transition. Increased activity of the calcium-dependent phosphatase calcineurin has been detected in cells with PINK RNAi, resulting in Drp1 dephosphorylation and increased GTPase activity, which may lead to enhanced fission. In addition, the disrupted mitochondrial calcium homeostasis in PINK1-deficient neurons may block mitochondrial transport in neurons through calcium-dependent inhibition of Miro. Upregulation of autophagy as well as increased mitochondrial biogenesis and respiratory complex synthesis have been observed in neurons lacking PINK1, and proposed to be protective compensatory mechanisms. Fragmented mitochondria have also been found frequently, though not always, in PINK1−/− cells. Mitochondrial fragmentation may serve a protective function by facilitating the degradation of dysfunctional mitochondria, especially in conjunction with increased autophagy. However, excessive fission may cause apoptosis. For details and references, please see main text
PINK1 and Parkin cooperate in the elimination of dysfunctional mitochondria and unfolded proteins
Flies lacking Parkin and PINK1 showed strikingly similar phenotypes, including dopaminergic neuron loss, apoptotic muscle degeneration and severe morphological defects of the mitochondria. While Parkin was able to rescue all phenotypes of PINK1-deficient flies, PINK1 was ineffective in complementing the defects in Parkin−/− flies [162, 163, 183]. Overexpression of Parkin also rescues mitochondrial fragmentation in PINK1 knockdown cells [148, 152, 157], but not vice versa [157]. These results establish that PINK1 and Parkin function in a common pathway to maintain mitochondrial integrity and dynamics, with Parkin acting downstream of PINK1.
Parkin may rescue the mitochondrial defects in PINK1−/− flies and cells by various ways, and the stimulation of autophagocytic degradation of functionally impaired mitochondria is likely one of them. Autophagy is an intracellular, lysosome-mediated degradation pathway for damaged proteins, organelles and membranes [184]. Autophagy is essential for neuronal survival, as shown by the fact that mice with neuron-specific deletions of the core autophagy genes Atg5 and Atg7 develop neurodegeneration, progressive deficits in motor function, accumulation of ubiquitin-positive inclusions and premature death [185, 186]. It has been demonstrated that Parkin is selectively recruited to dysfunctional mitochondria with low Δψm, promoting their engulfment and destruction in autophagosomes [187, 188]. This suggests that impaired removal of defective mitochondria may underlie the pathogenesis of AR-JP and possibly sporadic PD. Accordingly, a major function of Parkin is to stimulate mitophagy, and it has been proposed that Parkin may ubiquitinate as yet unknown mitochondrial proteins [189], thereby targeting mitochondria to autophagosomes in a manner similar to ubiquitin-tagged peroxisomes [190]. However, further studies with cells expressing ligase-deficient Parkin only, and identification of mitochondrial Parkin substrates will be necessary to confirm that ubiquitination plays a role in this process.
A recent overexpression study in human neuroblastoma cells revealed that PINK1 controls the translocation of Parkin from the cytosol to mitochondria through direct phosphorylation of Parkin on threonine 175 [145]. Recruitment of Parkin to the mitochondrial surface depended on the PINK1 kinase activity, implicating phosphorylation as an obligatory step [145]. In contrast, PINK1 gene silencing led to significantly lower amounts of Parkin in the mitochondria, and endogenous Parkin levels were also dramatically reduced in mitochondria from PINK1-deficient Drosophila compared to normal flies [145]. Phosphorylation of Parkin by PINK1 is conserved in flies (threonine 187 in Drosophila Parkin) and necessary for full rescue of the mitochondrial deficit in Parkin-deficient flies [145], suggesting that PINK1-mediated recruitment of Parkin to mitochondria serves a protective function. However, it is unlikely that PINK1-dependent redistribution of Parkin to mitochondria also enhances mitophagy, because overexpression of PINK1 suppressed injury-induced autophagy [152]. Therefore, phosphorylated Parkin in mitochondria likely has other roles. Possible functions of phosphorylated Parkin include replacing the degraded mitochondria by enhancing mitochondrial biogenesis [120] and/or limiting mitochondrial damage through the stimulation of mitochondrial DNA repair, replication and transcription [125] (Fig. 3). One possibility is that the PINK1 kinase activity towards Parkin increases upon activation of certain stress signaling pathways, resulting in enhanced recruitment of Parkin to mitochondria under conditions that might harm these organelles. This in turn could allow Parkin to repair damaged mitochondria and replace discarded organelles by new ones. Consistent with this idea, it has been shown that the activation of p38 kinase enhances PINK1-mediated phosphorylation of HtrA2 [144]. In the absence of PINK1, no phosphorylated Parkin would reach the mitochondria in stressed cells, potentially causing the propagation of faulty organelles. However, the reduced Δψm in PINK1−/− cells may trigger enough (unphosphorylated) Parkin to associate with defective mitochondria [187], resulting in their autophagocytic destruction. Nonetheless, the autophagy system would ultimately be overwhelmed by the mass of defective mitochondria, and neuron death would ensue.

Model for the cooperation of PINK1 and Parkin in the regulation of mitochondrial integrity and turnover, and the degradation of cytosolic aggregation-prone proteins. Cytosolic Parkin is effectively recruited to the surface of mitochondria with reduced Δψm, promoting the destruction of functionally impaired mitochondria in autophagosomes. PINK1 phosphorylates Parkin on Thr175, which results in redistribution of Parkin from the cytosol to mitochondria. PINK1-defcient cells and Drosophila have lower amounts of mitochondria-associated Parkin, suggesting that PINK1 physiologically increases the mitochondrial Parkin pool. The function of phosphorylated Parkin in mitochondria is not known. It is unlikely to be involved in mitophagy but, based on recent findings, may serve to stimulate the repair, replication and transcription of mtDNA, or to replace degraded organelles through mitochondrial biogenesis. A cytosolic E3 ubiquitin ligase complex containing PINK1, Parkin and DJ-1 has been implicated in the ubiquitination and degradation of Parkin substrates and unfolded/misfolded proteins that accumulate during heat shock, including Synphilin-1 and Parkin. Ablation of either PINK1 or DJ-1 reduced the proteasome-mediated degradation of these substrates, while overexpression of wildtype PINK1 but not PD-mutant PINK1 enhanced it. It has been suggested that the function of the PPD complex is to degrade potentially toxic proteins that accumulate and aggregate during oxidative stress. For details and references, please see main text
Impaired autophagy may indeed be central to PD pathogenesis [191], because several other PD genes interact with autophagy. Alpha-synuclein, which aggregates in Lewy bodies in sporadic and dominant familial PD [192–194], is degraded by both macroautophagy and chaperone-mediated autophagy (CMA) [195]. Familial PD mutants of α-synuclein bind to a lysosomal cargo receptor, thereby inhibiting their own degradation as well as the degradation of other CMA substrates in lysosomes [196]. In addition, DJ-1, linked to recessive EOPD [197], has been implicated in paraquat-induced autophagy [198].
CMA is particularly important for the degradation of cytosolic proteins [199] and, in conjunction with ubiquitin-dependent protein degradation by the proteasome, serves to prevent the accumulation and aggregation of misfolded and toxic proteins in cells. Recently, an ubiquitin E3 ligase complex comprised of Parkin, PINK1 and DJ-1 (termed PPD complex) has been identified in cells and human brain lysates, and shown to promote the degradation of unfolded proteins and Parkin substrates, such as Synphilin-1 [200]. Overexpression of PINK1 in neuroblastoma cells significantly enhanced Parkin-mediated degradation of heat shock-induced misfolded proteins, while genetic ablation of either PINK1 or DJ-1 resulted in increased accumulation of Parkin substrates in heat-shocked, but not normal, neurons [200]. In addition, both PINK1 and Parkin suppressed the toxicity of α-synuclein in cell and animal models of PD [201–204]. Collectively, these results suggest yet another mechanism by which PINK1 and Parkin cooperate to confer neuroprotection, namely the degradation of aggregation-prone, potentially toxic cytosolic proteins (Fig. 3). Finally, some of the Parkin substrates are known to aggregate and, at least one, the Pael receptor induces autophagy when overexpressed [205]. Therefore, inhibiting the accumulation of aggregation-prone proteins could prevent saturation of autophagy under conditions that enhance protein aggregation, such as oxidative stress in PD. Although a small amount of the PPD complex was found in the mitochondrial fraction in cells overexpressing the three recessive PD proteins, it is unclear whether the PPD complex also ubiquitinates mitochondrial proteins. Ubiquitination plays an important role in the regulation of mitochondrial dynamics [33, 206]. However, none of the currently known Parkin substrates are mitochondrial proteins, but additional studies addressing this possibility may be warranted.
Is there a link between PINK1, mitochondrial transport and dopamine neurotransmission?
As described earlier, Miro is an important calcium-sensitive regulator of axonal mitochondrial transport. In addition, overexpression of Miro affects mitochondrial morphology, at least in part by suppressing Drp1-mediated fission, resulting in mitochondrial thread formation and elongated mitochondria that more readily reached the dendrites in primary cortical neurons [65]. In transfected cells, PINK1 forms a multiprotein complex with Miro and Milton, suggesting that PINK1 may control mitochondrial transport and morphology via regulation of Miro and/or Milton [171]. Moreover, dysfunctional Ca2+ homeostasis in PINK1-deficient neurons and impaired calcium storage capacity of PINK1-deficient mitochondria [151] may cause the disruption of the Miro-KIF5 interaction, thereby blocking mitochondrial transport and energy supply in axons and synapses [63, 64]. Further experiments are necessary to study whether a defect of axonal mitochondrial transport does in fact exist in neurons lacking PINK1. If so, such a defect might explain the abnormal dopamine neurotransmission in mice lacking PINK1 [166], because mitochondrial dynamics and axonal trafficking of mitochondria are essential for synaptic function and neurotransmission [47–50].
Characteristics that may render the dopaminergic system particularly vulnerable to abnormal mitochondrial function and dynamics
Several features of the nigrostriatal system and the physiology of dopaminergic neurons may help to explain why dopaminergic neurons are particularly sensitive to perturbations in mitochondrial function and dynamics. For example, it has been shown that the mitochondrial density and mass are lower in the substantia nigra dopaminergic neurons, which preferentially degenerate in PD, compared to non-dopaminergic neurons or dopaminergic neurons of the ventral tegmental area that are spared from death in PD [207]. In addition, cytoplasmic dopamine is prone to auto-oxidation and enzymatic oxidation, which can lead to impaired mitochondrial respiration and membrane permeability [208, 209]. A proteomic study recently showed that several chaperones playing important roles in mitochondrial protein quality control and cristae organization, as well as proteins of the electron transport chain and enzymes of the matrix were covalently modified by dopamine, after exposure of rat brain mitochondria to dopamine quinone (DAQ) in vitro [210]. In addition, dopamine was shown to enhance mtNOS activity [211] and increase the susceptibility of PC12 cells to nitric oxide-dependent inhibition of respiration [212]. Dopamine also inhibited mitochondrial complex I-driven respiration when added short-term or long-term to the medium of cultured SH-SY5Y cells [213]. Moreover, administration of exogenous dopamine to hippocampal neurons blocked the trafficking of mitochondria [214]. Finally, dopamine has been shown to interfere with CMA [215] and to promote autophagy-dependent cell death via upregulation of α-synuclein expression [216]. Therefore, if not sequestered efficiently into synaptic vesicles, dopamine may affect mitochondrial respiration and transport. In addition, through its effects on autophagy, dopamine may interfere with autophagy-dependent clearance of aggregated proteins and dysfunctional mitochondria. Importantly, dopamine has been shown to covalently modify Parkin in living dopaminergic cells, which led to reduced Parkin solubility and inactivation of the Parkin E3 ligase activity [217]. Catechol-modified Parkin was specifically detected in the substantia nigra but not in other brain regions of PD patients, suggesting that dopamine-dependent inactivation of Parkin contributes to dopaminergic neuron loss [217]. Because Parkin is important for mitochondrial function and stimulates the clearance of dysfunctional organelles through mitophagy, dopamine-dependent Parkin inhibition may lead to mitochondrial dysfunction and impaired mitophagy. Collectively, these results show that dopamine and its metabolites can negatively affect mitochondrial homeostasis, rendering dopaminergic neurons more susceptible to further impairments of mitochondrial function, dynamics and clearance.
In addition, high levels of clonally expanded mtDNA deletions were shown to be present in individual pigmented neurons of the substantia nigra in aged people and PD patients [218, 219]. Because mitochondrial fission and fusion inhibit the propagation of mutant mtDNA genomes [178, 180, 220], abnormalities in mitochondrial dynamics could result in an expansion of mitochondria harboring mutant genomes in dopaminergic neurons, causing energy depletion and cell death.
Finally, a common feature of the neuronal populations that degenerate in PD is that they all have disproportionally long and thin axons in relation to their cell body [221]. This might pose a challenge for neurons, because they have to transport mitochondria over long distances in the axons, and the narrow diameter of these processes may be particularly sensitive to alterations in mitochondrial shape and size that could interfere with mitochondrial trafficking. It is also believed that the neuronal degeneration in PD starts at the axon terminals [221], which would be affected first by impaired mitochondrial dynamics due to their increased energy requirement and the strict dependence of synaptic function on mitochondria [47].
Conclusions
Significant progress has been made over the past decade in our understanding of the mechanisms and pathways that govern mitochondrial function and dynamics, and how pathogenic alterations of these mechanisms may contribute to the development of several neurodegenerative disorders, including autosomal dominant optic atrophy and Charcot-Marie-Tooth disease [222, 223], PD [101, 102] and Alzheimer’s disease [224, 225]. With relevance to PD, PINK1 maintains mitochondrial integrity through the regulation of mitochondrial membrane potential, cristae structure and calcium homeostasis. In addition, via direct phosphorylation of its mitochondrial substrates TRAP1 and HtrA2, PINK1 likely regulates mitochondrial protein quality control and proteolysis under conditions of stress.
Parkin is important for the repair of damaged and mutated mtDNA, the replication and transcription of the mitochondrial genome, as well as mitochondrial biogenesis. In addition, Parkin specifically eliminates dysfunctional mitochondria by promoting their degradation in autophagosomes. This may be particularly important in cells lacking PINK1, which accumulate mitochondria with impaired Δψm, and may explain how Parkin overexpression rescued the mitochondrial defects in PINK1-deficient flies and cells. In addition, the coordinated induction of fission and autophagy observed in PINK1-deficient neurons likely represents a compensatory response, which serves to fragment morphologically abnormal mitochondria and enhance their degradation in lysosomes (mitophagy). Finally, PINK1, Parkin and DJ-1 act in a complex to stimulate ubiquitin-dependent proteasomal degradation of aggregation-prone proteins, thus preventing the accumulation of potentially neurotoxic proteins. Understanding these mechanisms of injury and compensation in PINK1-deficient neurons is critical for the development of future therapies for PD and other neurodegenerative disorders caused by abnormal mitochondrial function and dynamics.
References
Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60:748–766
Elstner M, Andreoli C, Klopstock T, Meitinger T, Prokisch H (2009) The mitochondrial proteome database: MitoP2. Methods Enzymol 457:3–20
Bolender N, Sickmann A, Wagner R, Meisinger C, Pfanner N (2008) Multiple pathways for sorting mitochondrial precursor proteins. EMBO Rep 9:42–49
Westermann B (2008) Molecular machinery of mitochondrial fusion and fission. J Biol Chem 283:13501–13505
Hoppins S, Lackner L, Nunnari J (2007) The machines that divide and fuse mitochondria. Annu Rev Biochem 76:751–780
McBride HM, Neuspiel M, Wasiak S (2006) Mitochondria: more than just a powerhouse. Curr Biol 16:R551–R560
Cerveny KL, Tamura Y, Zhang Z, Jensen RE, Sesaki H (2007) Regulation of mitochondrial fusion and division. Trends Cell Biol 17:563–569
Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125:1241–1252
Chan DC (2006) Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22:79–99
Soubannier V, McBride HM (2009) Positioning mitochondrial plasticity within cellular signaling cascades. Biochim Biophys Acta 1793:154–170
Suen DF, Norris KL, Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22:1577–1590
Pitts KR, Yoon Y, Krueger EW, McNiven MA (1999) The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol Biol Cell 10:4403–4417
Yoon Y, Pitts KR, McNiven MA (2001) Mammalian dynamin-like protein DLP1 tubulates membranes. Mol Biol Cell 12:2894–2905
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256
Serasinghe MN, Yoon Y (2008) The mitochondrial outer membrane protein hFis1 regulates mitochondrial morphology and fission through self-interaction. Exp Cell Res 314:3494–3507
James DI, Parone PA, Mattenberger Y, Martinou JC (2003) hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278:36373–36379
Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M (2005) A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 16:5077–5086
Lee S, Jeong SY, Lim WC et al (2007) Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem 282:22977–22983
Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23:5409–5420
Yu T, Fox RJ, Burwell LS, Yoon Y (2005) Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J Cell Sci 118:4141–4151
Karbowski M, Jeong SY, Youle RJ (2004) Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol 166:1027–1039
Santel A, Frank S (2008) Shaping mitochondria: the complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB Life 60:448–455
Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282:11521–11529
Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8:939–944
Chang CR, Blackstone C (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282:21583–21587
Cereghetti GM, Stangherlin A, Martins de Brito O et al (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105:15803–15808
Chang CR, Blackstone C (2007) Drp1 phosphorylation and mitochondrial regulation. EMBO Rep 8:1088–1089
Cribbs JT, Strack S (2009) Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol 457:231–253
Harder Z, Zunino R, McBride H (2004) Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol 14:340–345
Zunino R, Braschi E, Xu L, McBride HM (2009) Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem 284:17783–17795
Zunino R, Schauss A, Rippstein P, Andrade-Navarro M, McBride HM (2007) The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J Cell Sci 120:1178–1188
Braschi E, Zunino R, McBride HM (2009) MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep 10:748–754
Neutzner A, Benard G, Youle RJ, Karbowski M (2008) Role of the ubiquitin conjugation system in the maintenance of mitochondrial homeostasis. Ann N Y Acad Sci 1147:242–253
Neuspiel M, Schauss AC, Braschi E et al (2008) Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18:102–108
Yonashiro R, Ishido S, Kyo S et al (2006) A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J 25:3618–3626
Karbowski M, Neutzner A, Youle RJ (2007) The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol 178:71–84
Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S (2006) MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep 7:1019–1022
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200
Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305:858–862
Chen H, Chomyn A, Chan DC (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280:26185–26192
Meeusen S, DeVay R, Block J et al (2006) Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 127:383–395
Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101:15927–15932
Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM (2004) Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem 279:18792–18798
Misaka T, Murate M, Fujimoto K, Kubo Y (2006) The dynamin-related mouse mitochondrial GTPase OPA1 alters the structure of the mitochondrial inner membrane when exogenously introduced into COS-7 cells. Neurosci Res 55:123–133
Sesaki H, Dunn CD, Iijima M et al (2006) Ups1p, a conserved intermembrane space protein, regulates mitochondrial shape and alternative topogenesis of Mgm1p. J Cell Biol 173:651–658
Fox EJ, Stubbs SA, Kyaw Tun J, Leek JP, Markham AF, Wright SC (2004) PRELI (protein of relevant evolutionary and lymphoid interest) is located within an evolutionarily conserved gene cluster on chromosome 5q34–q35 and encodes a novel mitochondrial protein. Biochem J 378:817–825
Chen H, Chan DC (2006) Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol 18:453–459
Keating DJ (2008) Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J Neurochem 104:298–305
Chang DT, Reynolds IJ (2006) Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol 80:241–268
Kang JS, Tian JH, Pan PY et al (2008) Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132:137–148
Morris RL, Hollenbeck PJ (1993) The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci 104:917–927
David G, Barrett EF (2003) Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol 548:425–438
Guo X, Macleod GT, Wellington A et al (2005) The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47:379–393
Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ (2005) Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47:365–378
Hollenbeck PJ (2005) Mitochondria and neurotransmission: evacuating the synapse. Neuron 47:331–333
Garcia-Chacon LE, Nguyen KT, David G, Barrett EF (2006) Extrusion of Ca2+ from mouse motor terminal mitochondria via a Na+–Ca2+ exchanger increases post-tetanic evoked release. J Physiol 574:663–675
Kann O, Kovacs R (2007) Mitochondria and neuronal activity. Am J Physiol Cell Physiol 292:C641–C657
Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL (2002) Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36:1063–1077
Tong JJ (2007) Mitochondrial delivery is essential for synaptic potentiation. Biol Bull 212:169–175
Hirokawa N, Takemura R (2005) Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 6:201–214
Glater EE, Megeath LJ, Stowers RS, Schwarz TL (2006) Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 173:545–557
MacAskill AF, Brickley K, Stephenson FA, Kittler JT (2009) GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol Cell Neurosci 40:301–312
Reis K, Fransson A, Aspenstrom P (2009) The Miro GTPases: at the heart of the mitochondrial transport machinery. FEBS Lett 583:1391–1398
Macaskill AF, Rinholm JE, Twelvetrees AE et al (2009) Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61:541–555
Saotome M, Safiulina D, Szabadkai G et al (2008) Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA 105:20728–20733
Liu X, Hajnoczky G (2009) Ca(2+)-dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int J Biochem Cell Biol 41:1972–1976
Jeyaraju DV, Cisbani G, Pellegrini L (2008) Calcium regulation of mitochondria motility and morphology. Biochim Biophys Acta 1787:1363–1373
Cai Q, Pan PY, Sheng ZH (2007) Syntabulin-kinesin-1 family member 5B-mediated axonal transport contributes to activity-dependent presynaptic assembly. J Neurosci 27:7284–7296
Cai Q, Gerwin C, Sheng ZH (2005) Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J Cell Biol 170:959–969
Li W, Bengtson MH, Ulbrich A et al (2008) Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS One 3:e1487
Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15:5001–5011
Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H (2005) Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem 280:25060–25070
Parone PA, James DI, Da Cruz S et al (2006) Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol 26:7397–7408
Parone PA, Martinou JC (2006) Mitochondrial fission and apoptosis: an ongoing trial. Biochim Biophys Acta 1763:522–530
Cassidy-Stone A, Chipuk JE, Ingerman E et al (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14:193–204
Tanaka A, Youle RJ (2008) A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol Cell 29:409–410
Sheridan C, Delivani P, Cullen SP, Martin SJ (2008) Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell 31:570–585
James DI, Martinou JC (2008) Mitochondrial dynamics and apoptosis: a painful separation. Dev Cell 15:341–343
Karbowski M, Lee YJ, Gaume B et al (2002) Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 159:931–938
Wasiak S, Zunino R, McBride HM (2007) Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol 177:439–450
Olichon A, Baricault L, Gas N et al (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278:7743–7746
Sugioka R, Shimizu S, Tsujimoto Y (2004) Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279:52726–52734
Scorrano L, Ashiya M, Buttle K et al (2002) A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2:55–67
Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C (2005) Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem 280:35742–35750
Frezza C, Cipolat S, Martins de Brito O et al (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126:177–189
Yamaguchi R, Lartigue L, Perkins G et al (2008) Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell 31:557–569
Germain M, Mathai JP, McBride HM, Shore GC (2005) Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J 24:1546–1556
Zhang D, Lu C, Whiteman M, Chance B, Armstrong JS (2008) The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. J Biol Chem 283:3476–3486
Delettre C, Griffoin JM, Kaplan J et al (2001) Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet 109:584–591
Guillery O, Malka F, Landes T et al (2008) Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell 100:315–325
Olichon A, Elachouri G, Baricault L, Delettre C, Belenguer P, Lenaers G (2007) OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ 14:682–692
Cipolat S, Rudka T, Hartmann D et al (2006) Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126:163–175
Pellegrini L, Scorrano L (2007) A cut short to death: Parl and Opa1 in the regulation of mitochondrial morphology and apoptosis. Cell Death Differ 14:1275–1284
Merkwirth C, Dargazanli S, Tatsuta T et al (2008) Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev 22:476–488
Merkwirth C, Langer T (2009) Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta 1793:27–32
Duvezin-Caubet S, Jagasia R, Wagener J et al (2006) Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem 281:37972–37979
Ishihara N, Jofuku A, Eura Y, Mihara K (2003) Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem Biophys Res Commun 301:891–898
Griparic L, Kanazawa T, van der Bliek AM (2007) Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol 178:757–764
Perkins G, Bossy-Wetzel E, Ellisman MH (2009) New insights into mitochondrial structure during cell death. Exp Neurol 218:183–192
Henchcliffe C, Beal MF (2008) Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol 4:600–609
Bueler H (2009) Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp Neurol 218:235–246
Van Laar VS, Berman SB (2009) Mitochondrial dynamics in Parkinson’s disease. Exp Neurol 218:247–256
Kitada T, Asakawa S, Hattori N et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Abbas N, Lucking CB, Ricard S et al (1999) A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s disease genetics study group and the European consortium on genetic susceptibility in Parkinson’s disease. Hum Mol Genet 8:567–574
Shimura H, Hattori N, Kubo S et al (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25:302–305
Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM (2000) Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA 97:13354–13359
Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105:891–902
Corti O, Hampe C, Koutnikova H et al (2003) The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: linking protein biosynthesis and neurodegeneration. Hum Mol Genet 12:1427–1437
Murakami T, Shoji M, Imai Y et al (2004) Pael-R is accumulated in lewy bodies of Parkinson’s disease. Ann Neurol 55:439–442
Periquet M, Corti O, Jacquier S, Brice A (2005) Proteomic analysis of parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J Neurochem 95:1259–1276
Fukae J, Sato S, Shiba K et al (2009) Programmed cell death-2 isoform1 is ubiquitinated by parkin and increased in the substantia nigra of patients with autosomal recessive Parkinson’s disease. FEBS Lett 583:521–525
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100:4078–4083
Pesah Y, Pham T, Burgess H et al (2004) Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131:2183–2194
Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ (2005) Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum Mol Genet 14:799–811
Palacino JJ, Sagi D, Goldberg MS et al (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279:18614–18622
Goldberg MS, Fleming SM, Palacino JJ et al (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635
Itier JM, Ibanez P, Mena MA et al (2003) Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet 12:2277–2291
Flinn L, Mortiboys H, Volkmann K, Koster RW, Ingham PW, Bandmann O (2009) Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 132:1613–1623
Mortiboys H, Thomas KJ, Koopman WJ et al (2008) Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol 64:555–565
Kuroda Y, Mitsui T, Kunishige M et al (2006) Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet 15:883–895
Wang C, Lu R, Ouyang X et al (2007) Drosophila overexpressing parkin R275 W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J Neurosci 27:8563–8570
Sang TK, Chang HY, Lawless GM et al (2007) A Drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J Neurosci 27:981–992
Lu XH, Fleming SM, Meurers B et al (2009) Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J Neurosci 29:1962–1976
Kao SY (2009) Regulation of DNA repair by parkin. Biochem Biophys Res Commun 382:321–325
Rothfuss O, Fischer H, Hasegawa T et al (2009) Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet 18:3832–3850
Valente EM, Abou-Sleiman PM, Caputo V et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Hatano Y, Li Y, Sato K et al (2004) Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol 56:424–427
Bonifati V, Rohe CF, Breedveld GJ et al (2005) Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65:87–95
Hedrich K, Hagenah J, Djarmati A et al (2006) Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: role of a single hit? Arch Neurol 63:833–838
Prestel J, Gempel K, Hauser TK et al (2008) Clinical and molecular characterisation of a Parkinson family with a novel PINK1 mutation. J Neurol 255:643–648
Rogaeva E, Johnson J, Lang AE et al (2004) Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol 61:1898–1904
Gelmetti V, Ferraris A, Brusa L et al (2008) Late onset sporadic Parkinson’s disease caused by PINK1 mutations: clinical and functional study. Mov Disord 23:881–885
Silvestri L, Caputo V, Bellacchio E et al (2005) Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 14:3477–3492
Beilina A, Van Der Brug M, Ahmad R et al (2005) Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci USA 102:5703–5708
Taymans JM, Van den Haute C, Baekelandt V (2006) Distribution of PINK1 and LRRK2 in rat and mouse brain. J Neurochem 98:951–961
Gandhi S, Muqit MM, Stanyer L et al (2006) PINK1 protein in normal human brain and Parkinson’s disease. Brain 129:1720–1731
Blackinton JG, Anvret A, Beilina A, Olson L, Cookson MR, Galter D (2007) Expression of PINK1 mRNA in human and rodent brain and in Parkinson’s disease. Brain Res 1184:10–16
Takatori S, Ito G, Iwatsubo T (2008) Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett 430:13–17
Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ (2008) Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet 17:602–616
Lin W, Kang UJ (2008) Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem 106:464–474
Zhou C, Huang Y, Shao Y et al (2008) The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci USA 105:12022–12027
Pridgeon JW, Olzmann JA, Chin LS, Li L (2007) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol 5:e172
Moriwaki Y, Kim YJ, Ido Y et al (2008) L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci Res 61:43–48
Plun-Favreau H, Klupsch K, Moisoi N et al (2007) The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol 9:1243–1252
Kim Y, Park J, Kim S et al (2008) PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun 377:975–980
Martins LM, Morrison A, Klupsch K et al (2004) Neuroprotective role of the reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol 24:9848–9862
Xu L, Voloboueva LA, Ouyang Y, Emery JF, Giffard RG (2009) Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab 29:365–374
Exner N, Treske B, Paquet D et al (2007) Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 27:12413–12418
Wood-Kaczmar A, Gandhi S, Yao Z et al (2008) PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 3:e2455
Sandebring A, Thomas KJ, Beilina A et al (2009) Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One 4:e5701
Gandhi S, Wood-Kaczmar A, Yao Z et al (2009) PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 33:627–638
Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284:13843–13855
Gegg ME, Cooper JM, Schapira AH, Taanman JW (2009) Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE 4:e4756
Haque ME, Thomas KJ, D’Souza C et al (2008) Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci USA 105:1716–1721
Deng H, Jankovic J, Guo Y, Xie W, Le W (2005) Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y. Biochem Biophys Res Commun 337:1133–1138
Petit A, Kawarai T, Paitel E et al (2005) Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J Biol Chem 280:34025–34032
Lutz AK, Exner N, Fett ME et al (2009) Loss of parkin or PINK1 function increases DRP1-dependent mitochondrial fragmentation. J Biol Chem 284:22938–22951
Liu W, Vives-Bauza C, Acin-Perez R et al (2009) PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson’s disease. PLoS ONE 4:e4597
Hoepken HH, Gispert S, Morales B et al (2007) Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol Dis 25:401–411
Piccoli C, Sardanelli A, Scrima R et al (2008) Mitochondrial respiratory dysfunction in familiar parkinsonism associated with pink1 mutation. Neurochem Res 33:2565–2574
Yang Y, Ouyang Y, Yang L et al (2008) Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA 105:7070–7075
Clark IE, Dodson MW, Jiang C et al (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–1166
Park J, Lee SB, Lee S et al (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441:1157–1161
Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA 105:1638–1643
Deng H, Dodson MW, Huang H, Guo M (2008) The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA 105:14503–14508
Kitada T, Pisani A, Porter DR et al (2007) Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA 104:11441–11446
Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci USA 105:11364–11369
Gispert S, Ricciardi F, Kurz A et al (2009) Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One 4:e5777
Gerencser AA, Nicholls DG (2008) Measurement of instantaneous velocity vectors of organelle transport: mitochondrial transport and bioenergetics in hippocampal neurons. Biophys J 95:3079–3099
Miller KE, Sheetz MP (2004) Axonal mitochondrial transport and potential are correlated. J Cell Sci 117:2791–2804
Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ (2009) Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 48:2045–2052
John GB, Shang Y, Li L et al (2005) The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell 16:1543–1554
Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell 16:59–68
Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW, You H (2009) FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc Natl Acad Sci USA 106:5153–5158
Sedding DG (2008) FoxO transcription factors in oxidative stress response and ageing—a new fork on the way to longevity? Biol Chem 389:279–283
Twig G, Elorza A, Molina AJ et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446
Benard G, Bellance N, James D et al (2007) Mitochondrial bioenergetics and structural network organization. J Cell Sci 120:838–848
Parone PA, Da Cruz S, Tondera D et al (2008) Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 3:e3257
Detmer SA, Chan DC (2007) Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 8:870–879
Amati-Bonneau P, Valentino ML, Reynier P et al (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131:338–351
Hudson G, Amati-Bonneau P, Blakely EL et al (2008) Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 131:329–337
Chen H, McCaffery JM, Chan DC (2007) Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130:548–562
Yang Y, Gehrke S, Imai Y et al (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA 103:10793–10798
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93
Hara T, Nakamura K, Matsui M et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Komatsu M, Waguri S, Chiba T et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
McBride HM (2008) Parkin mitochondria in the autophagosome. J Cell Biol 183:757–759
Narendra D, Tanaka A, Suen DF, Youle RJ (2009) Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 5:706–708
Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J (2008) Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA 105:20567–20574
Irrcher I, Park DS (2009) Parkinson’s disease: to live or die by autophagy. Sci Signal 2:21
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in lewy bodies. Nature 388:839–840
Polymeropoulos MH, Lavedan C, Leroy E et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Kruger R, Kuhn W, Muller T et al (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18:106–108
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L (2008) Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 283:23542–23556
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295
Bonifati V, Rizzu P, van Baren MJ et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259
Gonzalez-Polo R, Niso-Santano M, Moran JM et al (2009) Silencing DJ-1 reveals its contribution in paraquat-induced autophagy. J Neurochem 109:889–898
Cuervo AM (2006) Autophagy in neurons: it is not all about food. Trends Mol Med 12:461–464
Xiong H, Wang D, Chen L et al (2009) Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest 119:650–660
Petrucelli L, O’Farrell C, Lockhart PJ et al (2002) Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36:1007–1019
Haywood AF, Staveley BE (2004) Parkin counteracts symptoms in a Drosophila model of Parkinson’s disease. BMC Neurosci 5:14
Yasuda T, Miyachi S, Kitagawa R et al (2007) Neuronal specificity of alpha-synuclein toxicity and effect of Parkin co-expression in primates. Neuroscience 144:743–753
Todd AM, Staveley BE (2008) Pink1 suppresses alpha-synuclein-induced phenotypes in a Drosophila model of Parkinson’s disease. Genome 51:1040–1046
Marazziti D, Di Pietro C, Golini E, Mandillo S, Matteoni R, Tocchini-Valentini GP (2009) Induction of macroautophagy by overexpression of the Parkinson’s disease-associated GPR37 receptor. FASEB J 23:1978–1987
Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E (2008) Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9:505–518
Liang CL, Wang TT, Luby-Phelps K, German DC (2007) Mitochondria mass is low in mouse substantia nigra dopaminergic neurons: implications for Parkinson’s disease. Exp Neurol 203:370–380
Berman SB, Hastings TG (1999) Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J Neurochem 73:1127–1137
Premkumar A, Simantov R (2002) Mitochondrial voltage-dependent anion channel is involved in dopamine-induced apoptosis. J Neurochem 82:345–352
Van Laar VS, Mishizen AJ, Cascio M, Hastings TG (2009) Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol Dis 34:487–500
Czerniczyniec A, Bustamante J, Lores-Arnaiz S (2007) Dopamine enhances mtNOS activity: implications in mitochondrial function. Biochim Biophys Acta 1767:1118–1125
Antunes F, Han D, Rettori D, Cadenas E (2002) Mitochondrial damage by nitric oxide is potentiated by dopamine in PC12 cells. Biochim Biophys Acta 1556:233–238
Brenner-Lavie H, Klein E, Ben-Shachar D (2009) Mitochondrial complex I as a novel target for intraneuronal DA: modulation of respiration in intact cells. Biochem Pharmacol 78:85–95
Chen S, Owens GC, Edelman DB (2008) Dopamine inhibits mitochondrial motility in hippocampal neurons. PLoS One 3:e2804
Martinez-Vicente M, Talloczy Z, Kaushik S et al (2008) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118:777–788
Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S (2003) Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res 73:341–350
LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ (2005) Dopamine covalently modifies and functionally inactivates parkin. Nat Med 11:1214–1221
Bender A, Krishnan KJ, Morris CM et al (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517
Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K (2006) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 38:518–520
Malena A, Loro E, Di Re M, Holt IJ, Vergani L (2009) Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum Mol Genet 18:3407–3416
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134
Herzig S, Martinou JC (2008) Mitochondrial dynamics: to be in good shape to survive. Curr Mol Med 8:131–137
Liesa M, Palacin M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89:799–845
Wang X, Su B, Lee HG et al (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29:9090–9103
Reddy PH (2009) Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp Neurol 218:286–292
Acknowledgments
I thank Dr. Ravi Akundi of the Department of Anatomy and Neurobiology, University of Kentucky for valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Büeler, H. Mitochondrial dynamics, cell death and the pathogenesis of Parkinson’s disease. Apoptosis 15, 1336–1353 (2010). https://doi.org/10.1007/s10495-010-0465-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-010-0465-0