
Abstract
Autosomal recessive polycystic kidney disease (ARPKD) is caused by mutations in PKHD1, a gene encoding fibrocystin/polyductin (FC1), a membrane-associated receptor-like protein involved in the regulation of tubular cell adhesion, proliferation and apoptosis. Although it is generally accepted that apoptosis is implicated in ARPKD, the question of whether increased apoptosis is a normal response to abnormal cell proliferation or, instead, it is a primary event, is still subject to debate. In support of the latter hypothesis, we hereby provide evidence that apoptosis occurs in the absence of hyper-proliferation of FC1-depleted kidney cells. In fact, a decrease in cell proliferation, with a concomitant increase in apoptotic index and caspase-3 activity was observed in response to FC1-depletion by PKHD1 siRNA silencing in HEK293 and 4/5 tubular cells. FC1-depletion also induced reduction in ERK1/2 kinase activation, upregulation of the pro-apoptotic protein p53 and activation of NF-κB, a transcription factor which reduces apoptosis in many organs and tissues. Interestingly, selective inactivation of NF-κB using either an NF-κB decoy or parthenolide, a blocker of IKK-dependent NF-κB activation, reduced, rather then increased, apoptosis and p53 levels in FC1-depleted cells. Therefore, the proapoptotic function of NF-κB during cell death by FC1-depletion in kidney cells is evident.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autosomal recessive polycystic kidney disease (ARPKD) is an important cause of renal and liver-related morbidity and mortality in neonates and infants [1, 2]. This disease is caused by mutations in the polycystic kidney and hepatic disease gene 1 (PKHD1) which encodes fibrocystin/polyductin (FC1), a ~ 500 kDa type I membrane protein expressed in the primary cilium and plasma membrane of renal and bile duct epithelial cells [3]. The structure and localization of FC1 suggest that it may function as a receptor for an, as yet, unidentified ligand which is involved in maintaining the 3-dimensional tubular architecture. FC1 may form a functional complex with polycystin-1 (PC1) and polycystin-2 (PC2), which have been observed to be mutated in autosomal dominant polycystic kidney disease (ADPKD) [4, 5]. In fact, FC1, as well as, polycystins themselves, are essential for the control of epithelial differentiation, proliferation and apoptosis which is mediated via the integration of complex signaling events. Apoptosis is central to normal kidney development [6] and has been implicated in kidney diseases of various origins, including PKD [7]. Indeed, the cystic epithelia in both ADPKD and ARPKD display de-differentiated cells with polarization defects, high rates of division and apoptosis [7–11].
To better define the elevated susceptibility to programmed cell death in ARPKD and the role therein of FC1, we investigated proliferation and apoptosis in human kidney cells possessing down-regulated PKHD1 due to siRNA interference. Our results demonstrate that there is an increase in apoptosis, which is dependent on caspase-3 activation and associated to abnormally elevated activity of the nuclear factor-kappa B (NF-κB), in FC1-deficient human cells. This is a transcription factor which is crucial for the regulation of many physiological and pathological-associated processes; it is closely associated with cancer development and it plays a pivotal role in promotion of cell proliferation and inflammation, as well as suppression of apoptosis [12]. Nevertheless, NF-κB activation is known to accompany apoptotic death induced by serum withdrawal in HEK293 cells [13]. Therefore, the activation of this transcription factor may be responsible for triggering a distinct pattern of target genes which induce diametrically opposite effects in different tissues or contexts [12, 14, 15]. Indeed, we have demonstrated that NF-κB inactivation is required for the suppression of caspase-3 activity and reduction of cell death in FC1-depleted cells, thus showing that NF-κB may play a pro-apoptotic role in polycystic cells.
Methods
Reagents
D-MEM/F12 and MEM media, fetal bovine serum (FBS), glutamine and antibiotics were obtained from Euroclone (Milano, Italy); G418, Tumor Necrosis Factor α (TNF), protease inhibitors, Bovine Serum Albumin (BSA), mouse anti-β-actin and anti-β-tubulin were from Sigma (Milano, Italy). Parthenolide was obtained from Alexis Biochemicals (Vinci, Italy). pNF-κB-TA-Luc from Clontech (BD bioscences, Buccinasco, Italy), scramble siRNA from Ambion (Applied Biosystems, Monza, Italy), the antibodies anti-p21(Waf), -NFκB-p65, -p53 were from Santa Cruz (DBA Italia Srl, Segrate, Italy), and anti-Erk1/2 and -pErk1/2 were from Cell Signaling Technology (Euroclone, Italy). Enhanced chemiluminescent substrates for immunoblotting (SuperSignalDura or SuperSignalFemto) and HRPconjugated goat anti-rabbit and anti-mouse antibodies were purchased from Pierce (Euroclone, Italy).
Cells, DNA constructs and transfections
Human embryonic (HEK293), and adult 4/5 kidney tubular cells (4/5) were cultured as previously described [16]. Cells were co-transfected by calcium phosphate with pCDNA3 and either pSsiPKHD1a or pSsiPKHD1b recombinant plasmids expressing PKHD1 siRNA, or the empty pSuper vector, respectively. PKHD1 siRNA a and b (against PKHD1 exon VI 5′GTTTATCCACCAAGTGGTG3′ and exon VIII 5′GCTCAAGGA GACAAATGGG3′ sequences, GeneBank accession number AF480064) were cloned as previously described [16]. G418-resistant clones (HEK293pSuper, HEK293pSsiPKHD1, 4/5pSuper and 4/5pSsiPKHD1), and cells transfected with scramble siRNA [16] were analysed by RT-PCR and immunoblotting.
RT-PCR analysis
Total RNA was purified with Trizol® (Invitrogen, San Giuliano Milanese, Italy). mRNA was reverse-transcribed using a IM-Prom II reverse transcriptase kit (Promega, Milano, Italy) with random hexamers and specific primers for FC1 PCR and nested PCR as already described [17].
Western blotting
Total cell lysates were obtained as described [16]. For the protein phosphorylation analysis, cell lysates were supplemented with 10 mM sodium pyrophosphate and 1 mM sodium vanadate. Lysates were subjected to 6 or 10% SDS–polyacrylamide gel electrophoresis. For cell membranes preparation [18, 19] cells were homogenized (Dounce) then centrifuged at 1,500g for 10 min; the supernatant was centrifuged at 15,000g for 1 h. Pellets were resuspended in lysis buffer, separated on SDS–polyacrylamide gel, blotted and analyzed as previously described [16]. The monoclonal antibody anti-FC1 14A N-ter was employed for FC1 detection [20] and proteins were visualized by using the SuperSignalFemto chemiluminescence system. Quantitative band analysis was performed by scanning filters with a Model GS-700 Imaging Densitometer (BIO-RAD, Milan, Italy).
Cell proliferation and cycle assays
Cells (30,000 cells/ml) were plated overnight in 10% FBS-supplemented medium in 24-well plates. After plating, cells were starved for 24 h in medium supplemented with 0.4% BSA, and then cultured in medium containing 1% FBS. Cell proliferation was calculated by direct counting after trypan blue staining, using a Burker chamber. For cell cycle analysis, cells (300,000/ml), in 6-well plates, were starved for 24 h in 0.4% BSA, and stimulated for an additional 24 h in 1% FBS. Analysis was carried out by flow cytometry of propidium iodide-treated cells, using the Becton–Dickinson FACSCalibur Immunocytometry System [16].
Apoptosis assays
Cells were cultured on 24-mm glass coverslips in 0.4% BSA for 24 h, and treated with 1% FBS or with the appropriate molecules and times as specifically indicated. For UV treatment, cells were exposed to 70 J/m2 UV radiation. After treatments, apoptotic cell presence was determined by fluorescence after staining with Hoechst 33258 (10 μg/ml). Images were acquired with a Zeiss Axiovert 200 fluorescence microscope, equipped with a back-illuminated CCD camera (Roper Scientific, Tucson, AZ), excitation and emission filter wheels (Sutter instrument company, Novato, CA) and piezoelectric motoring of the Z stage (Physik Instrument, GmbH and Co, Karlsruhe, Germany). Experiments were performed on stably transfected clones after at least 6 month from cloning.
Caspase-3 assay
Caspase-3 activity was evaluated using an EnzChek® caspase-3 Assay Kit (Invitrogen). After treatment with 1% FBS alone or in combination with the specified molecules for 24 h, cells were harvested, lysed and centrifuged according to the manufacturer’s instructions. Protein content of the lysates was quantified by the Bradford assay. About 50 μl aliquots of supernatant were incubated with 50 μl of 2X substrate working solution containing 5 mM Z-DEVD-R110. Fluorescence was measured at 520 nm by a fluorimeter (VICTOR3 1420 Multilabel Counter, PerkinElmer) every min for 90 min. Values were normalized to the amount of protein samples. Caspase activity of each cell sample was represented by the maximal increase as an average value of the final 10 min. Furthermore, caspase-9 activity was determined using the Caspase-Glo® 9 Assay (Promega).
NF-κB activation
NF-kB transcriptional activity was assayed by trasfection with a luciferase reporter plasmid as previously described [16]. Cells were seeded in 6-well plates, cultured to 80% confluence in 10% FBS and co-transfected with pNF-κB-TA-Luc and pCMV/β-galactosidase. After 8 h, cell medium was refreshed with a 0.4% BSA-supplemented medium for an additional 24 h, together with the appropriate molecules and at suitable times, as specifically indicated. Cells were then washed once with cold PBS and lysed for measurement of luciferase activity. Lysates were normalized for protein content. Data were expressed as relative luciferase units normalized by β-galactosidase units in n-fold variations with respect to control cells.
For the NF-κB-GFP localization assay, fluorescence analysis was performed on cells transfected with the NF-κB-GFP construct by using the Ca2+ phosphate procedure [19]. NF-κB subcellular localization was analysed using a digital fluorescence microscope (Zeiss Axiovert 200), and images were captured and recorded using a digital-imaging system [21].
Ca2+ measurement
Measurement of ATP-evoked calcium levels was performed in cells grown on coverslips and transfected with recombinant cytoplasmic aequorin (cyt-Aeq) cDNA [22]. Two hours after aequorin reconstitution, cells on coverslips were transferred into the perfusion chamber of a luminometer and continuously perfused at the rate of 2.5 ml/min which ensures rapid exchange of perfusate and cell adhesion to coverslips. Following 1 min of pefusion, cells were stimulated with 100 μM ATP, as previously reported [23]. Cytoplasmic Ca2+ was measured in Fura-2AM-loaded 4/5 cells, which are negligibly transfectable by cyt-Aeq cDNA [16]. Cells, grown on coverslips, were starved overnight with BSA, loaded with Fura-2AM (4 μM/30 min/37°C), and transferred to the thermostated stage of a Zeiss Axiovert 200 inverted microscope equipped with a Sutter filterwheel and 340/380 excitation filters. Cells were stimulated with 100 μM ATP for 15 min, and images were acquired at a frequency of 1 image/s. Signals were computed into relative ratio units of the fluorescence intensity at different wavelengths (340/380 nm). Calibration was carried out using MetaFluor software, according to a previously reported equation [23].
Statistical analysis
Data were presented as mean ± SD or SEM, and the statistical significance of differences among three groups was calculated by Anova followed by a Dunnett test. In addition, differences between two groups were calculated by the paired or unpaired Student’s t-test. P values < 0.05 were regarded as statistically significant.
Results
Effect of FC1 suppression on the cell cycle
PKHD1 RNA was undetectable in the majority of the clones stably transfected with either a or b PKHD1 siRNA-expressing plasmids (psiPKHD1, Fig. 1a, upper part). PKHD1-downregulated (HEK293pSsiPKHD1) clones were considered those that showed reduction or absence of the FC1 protein, as compared with those transfected with either the empty vector or scramble siRNA sequence (Fig. 1a, lower part). Similar findings were observed in kidney tubular 4/5 cells which were stably transfected with psiPKHD1a (Fig. 1a, inset). Selected clones (4/5pSsiPKHD1a1 and 4/5pSsiPKHD1a2) were found to be partially PKHD1-downregulated with ~40% FC1 in contrast with control 4/5pSuper.

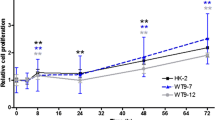
Reduced proliferation of FC1-depleted cells. a Downregulation of PKHD1 mRNA and FC1 protein in HEK293 and 4/5 (inset) cells stably transfected with either pSuper empty or PKHD1-siRNA a or b expressing plasmids. RT-PCR and Western blot analysis of membrane proteins were performed as described in “Methods”. Numbers above the blots stand for clone numbers. Blots were stripped and re-probed with β-tubulin, b, c PKHD1 silencing decreased cell proliferation. Cells were cultured with 1% FBS for up to 72 h. After 48 h culture, proliferation was analysed (right panels). Bars show average results (±SD) of various clones in duplicate experiments (n = 4 for HEKpSuper, HEKpSsiPKHD1a and HEKpSsiPKHD1b clones; n = 3 for 4/5pSuper and 4/5pSsiPKHD1a cells. Analysis was by ANOVA followed by Dunnett test in b and by Student’s t test in c. * P < 0.05, in silenced versus control clones)
FC1 depletion caused a reduction in cell proliferation of both HEK293pSsiPHKD1 and 4/5pSsiPHKD1 cells. As shown in Fig. 1b, cell proliferation in HEK293pSsiPHKD1 clones was approximately 25% below that of control clones (HEK293pSsiPHKD1a and HEK293pSsiPHKD1b vs. controls, P < 0.05). Since the reduction was observed in different a and b clones, subsequent experiments were performed with HEK293pSsiPHKD1a6 and HEK293pSsiPHKD1b14 clones, which yielded a similar 30% reduction. This reduction was also observed in transiently pSsiPKHD1-transfected, but not in scramble siRNA-transfected, HEK293 cells (Supplemental Fig. 1). The proliferation of 4/5pSsiPHKD1a cells was roughly 30% lower than in 4/5pSuper cells (Fig. 1c), and was also observed in transiently pSsiPKHD1a-transfected cells (P < 0.05) (Supplemental Fig. 1).
The percentage of cells in phase G1 was slightly increased in FC1-downregulated cells with respect to control cells. The ratio between G2/M and G1 percent values was lower in HEK293pSsiPHKD1 and 4/5pSsiPHKD1 than in HEK293pSuper and 4/5pSuper cells (Supplemental Fig. 2, right panels, P < 0.05). Therefore, FC1-downregulation was associated to a small reduction in the number of cycling cells, mainly in the transitions from phases G0/G1 to S, and from S to G2/M.
Effect of FC1 suppression on cell apoptosis
The number of dead cells, measured as Trypan blue positivity, was at least doubled in 1% FBS-treated 4/5pSsiPHKD1 and HEK293pSsiPHKD1 cells with respect to controls, even after transient expression of PKHD1 siRNA (Fig. 2a, left, central and right panel, respectively).

Increased apoptosis in FC1-depleted cells. a Trypan blue positivity in 4/5 and HEK293 clones after 48 h culture in 1% FBS. Bars show the mean ± SD of at least three experiments with 4/5pSuper, 4/5pSsiPKHD1a1 and 4/5pSsiPKHD1a2 (left panel), and in HEKpSuper, HEKpSsiPKHD1a and HEKpSsiPKHD1b (central panel). Analysis was by ANOVA followed by Dunnett test (** P < 0.01, *** P < 0.001). Results obtained in HEK293 cells 24 h after transient transfection are also shown (right panel), b Hoechst-33258 staining of nuclei after 24 h culture in 1% FBS. Bars show relative apoptotic index, calculated using the mean fluorescence value of groups of at least 50 cells on a coversleep, collected along 20 planes on the Z axes (Z-series) (see “Methods”) (** P < 0.01, Student’s t test for paired data, n = 5), c caspase-3 activity in HEK293 and 4/5 clones cultured for 24 h in 1% FBS (left panels) (see “Methods”). Bars (upper part) show mean ± SE. Analysis was by ANOVA followed by Dunnett test, (* P < 0.05, ** P < 0.01). Similarly, caspase-3 activity in 4/5pSsiPKHD1 is higher than in 4/5pSuper cells, also after UV-irradiation. Bars (lower part) show mean ± SD of three experiments; * P < 0.05 and ** P < 0.01 versus untreated and treated cells, respectively, Student’s t test
A twofold increase in the number of apoptotic nuclei was also observed after Hoechst staining in HEK293pSsiPHKD1 cells (Fig. 2b). The increase in Hoechst staining as well as in trypan blue positivity did not change in stable clones as time passed (data not shown). Therefore, FC1-depletion lowers cell viability by increasing the number of apoptotic nuclei.
Consistently, caspase-3 activity was higher in both HEK293pSsiPHKD1 and 4/5pSsiPHKD1 cells (Fig. 2c), and even more so after UV treatment (P < 0.01 in UV-treated 4/5pSsiPHKD1 versus UV-treated 4/5pSuper cells).
Furthermore, caspase 9 activity was elevated (approximately 1.6 fold) in HEK293pSsiPHKD1 clones (Supplemental Fig. 3), indicating that an induction of the intrinsic pathway of apoptosis takes place in FC1-depleted clones.
Effect of FC1 suppression on cell proliferation and apoptosis-related genes
FC1-depletion triggers modifications in the expression of genes related to cell proliferation and/or apoptosis in HEK293pSsiPHKD1 cells (Fig. 3a). In particular, levels of the proapoptotic mediator p53 were higher in HEK293pSsiPHKD1 than in HEK293pSuper cells (P < 0.01) in the absence of 1% FBS, and they were not further incremented by treatment with FBS. In contrast, levels of p21 were not significantly modified by FC1 depletion, either in the absence or presence of 1% FBS. Furthermore, 1% FBS treatment reduced the level of phosphorylated ERK1/2 kinase in HEK293pSsiPHKD1, but not in HEK293pSuper cells (Fig. 4b) (P < 0.01 in FBS-treated versus -untreated HEK293pSsiPHKD1 cells). Results indicate that FC1-depletion increases the expression or stability of p53 without affecting p21 expression, but reduces the FBS-dependent activation of ERK1/2 kinase.

Increased p53 expression and decreased ERK activation in HEKpSsiPKHD1 cells. a Immunoblotting analysis of p53 and p21 in HEKpSuper (pS) and HEKpSsiPKHD1 (pSsiPKHD1) cells that have been cultured for 12 h in the presence of 0.1% FBS and then for another 15 h with or without 1% FBS. Total cell lysates were analysed by 10% acrylamide SDS–PAGE, blotted and probed with anti-p53 and anti-p21 as described in “Methods”. Densitometric values of the protein bands were normalized for those of β-actin. Bars represent the mean ± SD of three experiments. ** P < 0.01 versus untreated HEKpSuper cells, b analysis of ERK activation in cells after 12 h culture with 0.4% BSA and 15 min stimulation with or without 1% FBS. Total lysates were probed with anti-phospho-ERK antibody and than re-probed with anti-ERK antibody. Density of phospho-ERK1/2 bands was normalized against that of ERK1/2. Bars represent the mean ± SD of P-ERK/ERK values related to untreated HEKpSuper cells. ** P < 0.01 compared to untreated HEKpSsiPKHD1 cells (paired data, n = 8)

Increased NF-κB activity in FC1-depleted cells. a NF-κB activation measured by luciferase promoter activity in NF-κB-pTA-luc-transfected HEK293 and 4/5 cells. Cells were transfected as described in “Methods”. NF-κB activation was determined by measuring the luciferase/β-galactosidase ratio (luc/β-gal) in stably pSsiPKHD1-transfected cells. Results represent the mean ± SD of at least three experiments. Analysis was by ANOVA followed by Dunnett test (*** P < 0.001 in FC1-depleted vs. control cells), b 24 h after transfection, cells were stimulated overnight with 15 ng/ml TNFα. Results represent the mean ± SD of at least three experiments. (** P < 0.01 in treated vs. untreated cells, Student’s t test), c immunoblot analysis of NF-κB p65 and β-tubulin in HEK293pSuper and HEK293pSsiPKD1 cells before NF-κB -pTA-luc transfection, d fluorescence of p65 NF-κB -GFP-transfected cells. Cells, grown on coverslips, were transfected for 16 h with the NF-κB-GFP construct, cultured for another 24 h in 1% FBS and analysed by fluorescence microscope as described in “Methods”. Arrows indicate prevalent and moderate NF-κB-GFP nuclear positivity in HEK293pSsiPKD1 cells. Magnification 60×. Bars show the nuclear positivity in HEK293pSuper (n = 52) and in HEK293pSsiPKD1 (n = 84) cells as % of total nuclei. *** P < 0.001, Student’s t test
Since FC1-depletion was associated to increased apoptosis and reduced cell proliferation, we examined the effect of FC1 deficiency on the activity of the transcription factor NF-κB, a regulator of these cell processes in the kidney and several other systems [24]. NF-κB-dependent luciferase-reporter activity was markedly increased in FC1-depleted cells (Fig. 4a): about sixfold higher in HEK293pSsiPHKD1, and fourfold higher in 4/5pSsiPHKD1 than in control cells (P < 0.001 and P < 0.01, respectively). Interestingly, the treatment with TNFα (15 ng/ml), a canonic activator of the NF-κB pathway, increased NF-κB activity in control, but not in HEK293pSsiPHKD1 cells (Fig. 4b, P < 0.05 versus untreated cells). FC1-depletion was, therefore, associated to an increase in NF-κB activity, not further incremented by treatment with TNFα.
Since levels of p65, a component of the NF-κB protein complex, did not differ between HEK293pSuper and HEK293pSsiPHKD1 cells (Fig. 4c), the observed increase in NF-kB activity was shown not to be caused by increased expression of p65, but by an increase in NF-κB activation. Consistently, nuclear fluorescence was variably detectable in HEK293pSsiPHKD1 cells transiently expressing a p65 NF-κB-GFP chimeric protein, but absent or very low in their HEK293pSuper counterparts (Fig. 4d). Overall nuclear positivity was increased (at least threefold) in HEK293pSsiPHKD1 cells.
Effects of NF-κB inhibitors on cell apoptosis
In order to verify whether a relationship between increased apoptosis and NF-κB activation exists in FC1-depleted cells, HEK293pSsiPHKD1 were incubated with 10 μM parthenolide (PN), a sesquiterpene lactone that results in blockade of IKK-dependent activation of NF-κB [25].
Pre-incubation with PN caused a reduction in NF-κB activity (P < 0.05 versus untreated cells) in HEK293pSsiPHKD1 cells (Fig. 5a). However, PN did not cause variations in cell growth (Fig. 5b) or death (Fig. 5c) in HEK293pSsiPHKD1 cells, while it did it in HEK293pSuper cells. Consistently, caspase-3 activity was increased in PN-treated HEK293pSuper cells (~1.3-fold, P < 0.05 versus untreated cells), but not in HEK293pSsiPHKD1 cells (Fig. 5d). Similarly, PN treatment increased caspase-3 activity in 4/5pSuper (~1.5-fold, P < 0.05), but not in 4/5pSsiPHKD1 cells) (Fig. 5, inset). FC1-depleted cells were, therefore, unresponsive to the apoptotic effect of PN. Thus, NF-κB may play an apoptotic rather than an anti-apoptotic role in these cells. Interestingly, in contrast to HEK293pSuper, PN reduced p53 levels (P < 0.01 versus untreated cells) in HEK293pSsiPHKD1 cells (Fig. 5e). Overall, apoptosis in these cells is likely to be NF-κB-dependent and mediated by p53.

Absence of parthenolide-dependent apoptosis in FC1-depleted cells. a NF-κB activity in PN-treated HEK293 cells. Cells, after 24 h transfection with NF-κB-pTA-luc, as described in “Methods”, were stimulated with PN (10 μM) for an additional 16 h. Results are expressed as n-fold activation of treated versus untreated cells, and reflect the mean ± SEM of at least three duplicate experiments. * P < 0.05, b, c cell proliferation and cell death were assessed by counting Trypan blue-negative and trypan blue-positive cells cultured overnight in serum-free medium and for an additional 8 h with PN. Results are expressed as n-fold variations of treated versus untreated cells (mean ± SD of three duplicate experiments; * P < 0.05), d caspase-3 activity in HEK293 and 4/5 cells (inset) cultured for 16 h with and without PN. Bars show the mean ± SD obtained in triplicate experiments. * P < 0.05, e p53 and β-actin levels measured by immunoblot in cells, as described in d and in Fig. 3 legend. Bars show the mean ± SD obtained in at least three experiments. ** P < 0.01 versus untreated cells
The possible pro-apoptotic role of NF-κB in FC1-depleted cells was further investigated by selective inactivation of NF-κB in HEK293 cells with an NF-κB decoy, a synthetic oligonucleotide (ODN) which can imitate the NF-κB binding site [26]. As expected, the ODN (10 ng/ml) caused a marked and comparable reduction of NF-κB-binding activity in both HEK293pSuper and HEK293pSsiPHKD1 cells (Fig. 6a). The decoy effect resulted in increased caspase-3 activity in HEK293pSuper cells, but in its reduction in HEK293pSsiPHKD1 cells (Fig. 6b): caspase-3 activity in ODN-treated, as compared to untreated cells, was found to be increased 1.43 ± 0.28-fold in HEK293pSuper and reduced 0.8 ± 0.14-fold in HEK293pSsiPHKD1 cells, P < 0.05. Therefore, the loss of NF-κB activity reduced the apoptotic features of FC1-depleted cells, consistent with a pro-apoptotic role of NF-κB in these cells.

NF-κB is required for FC1-depletion-dependent apoptosis. a Loss of NF-κB activity in NF-κB decoy-treated HEK293 cells. 24 h after transfection with NF-κB -pTA-luc (see “Methods”), cells were incubated with NF-κB ODN decoy (10 ng/ml) for an additional 24 h before luciferase activity measurement, b results of caspase-3 assays are expressed as n-fold variations of NF-κB decoy-treated cells versus untreated-cells (mean ± SD of three duplicate experiments; * P < 0.05)
Effect of FC1 suppression on intracellular Ca2+ concentration
Based on the functional interaction between FC1 and the polycystin-2 (PC2) Ca2+ channel [4], intracellular Ca2+ concentrations were compared in FC1-depleted and control cells. As expected from the FC1-dependent stability of PC2 [27], PC2 levels were lower in FC1-depleted cells (Fig. 7a). Consistent with the PC2-dependent reduction in Ca2+ release from ER [28], ATP-evoked Ca2+ levels were increased in HEK293pSsiPHKD1 and 4/5pSsiPHKD1 cells (Fig. 7b).

PC2 expression and ATP-evoked Ca2+ levels in FC1-depleted cells. a Immunoblot analysis of PC2 in HEK293 and 4/5 cells stably transfected with pSuper and pSsiPKHD1 plasmids. Filters were probed with anti-PC2 N-ter [21] and with anti-β-tubulin antibodies, as described in “Methods”, b representative traces of 100 μM ATP-evoked cytoplasmic Ca2+ levels in HEK293pSuper (black) and HEK293pSsiPKHD1 cells (grey). Ca2+ concentrations were measured in cyt-Aeq-transfected cells using a luminometer, as detailed in “Methods”. Results are expressed as n-fold increase in FC1-depleted versus control cells (mean ± SD of three duplicate experiments; ** P < 0.01), c 100 μM ATP-evoked Ca2+ levels in 4/5 cells transiently transfected with pSuper or pSsiPKHD1 b plasmids. Analysis was performed in Fura-2AM loaded cells by recording fluorescent images with a microscope, as described in “Methods”. Results are expressed as n-fold increase in FC1-depleted versus control cells (mean ± SD of three experiments; * P < 0.05)
Discussion
In PKD, cell apoptosis is one of several improperly functioning pathways involved in cyst formation [7, 11, 29–31]. In this study we demonstrate that FC1 is involved in the control of apoptosis through the modulation of NF-κB. In fact, in FC1-depleted cells, (1) basal and UV-induced caspase-3 activity is higher than in control cells; (2) expression of the pro-apoptotic tumor suppressor protein p53 is increased, in association with a fourfold increase in the activitation of NF-κB; (3) treatment with PN, which inhibits IKK-dependent NF-κB activation, does not increase the apoptosis which occurs in control cells; (4) selective inactivation of NF-κB using an NF-κB ODN decoy reduces caspase-3 activity which, instead, is increased in control cells. This observation is new for kidney cells, in which NF-κB has been reported as a potent anti-apoptotic response to a ubiquitous array of apoptotic triggers [25, 32, 33].
The fact that downregulation of PKHD1 renders embryonal HEK293 and adult 4/5 tubular kidney cells more susceptible to apoptosis is in agreement with the notion that apoptosis is implicated in ARPKD [11, 34, 35]. On the other hand, the association between the increased apoptosis and the reduced cell proliferation in FC1-depleted cells was unexpected for polycystic diseases which are associated to increased proliferation. Indeed, both of these cellular features appear to be PKHD1 siRNA-specific as they do not occur in scramble siRNA-treated cells, nor in cells that have been silenced by PKD1 siRNA-expressing plasmids, which showed, in contrast, increased cell proliferation without a rise in apoptosis [19]. Moreover, cell features observed in FC1-depleted cells are consistent with previous findings showing increased apoptosis and reduced proliferation in mouse renal tubular IMCD cells after Pkhd1-silencing [36]. These observations are in apparent contradiction to other findings [37] which have shown no differences in proliferation between control and PKHD1-silenced HEK293T cells. However, these cells were investigated after culture in 10% rather than 1% FBS.
Increased apoptosis and reduced cell proliferation in FC1-depleted cells, compared to wild type cells, are consistent with their increased expression of the pro-apoptotic tumor-suppressor protein p53, and with reduced activation of Erk1/2 (Fig. 3). This latter finding is in agreement with those reported in Pkhd1-silenced IMCD cells, where a reduction in Erk1/2 phosphorylation has been associated to that in FAK [36], a kinase proposed as a critical scaffold protein which sequesters pro-apoptotic proteins such as p53 to mediate cell survival [38]. The reduced activation of Erk1/2 may therefore have produced the increased expression of p53 in FC1-depleted cells. Interestingly, the reduction in p53 observed after NF-κB inhibition by PN in FC1-depleted cells (Fig. 5) also lends support to the concept that p53 expression is either dependent on or sustained by NF-κB activation in these cells. In this regard, it is well established that NF-κB activation could increase p53 levels [39] and that p53 could activate NF-κB transcriptional activity, thereby contributing to apoptosis [40]. In fact, loss of NF-κB, has been shown to abrogate p53-induced apoptosis, thus implying that it is essential for p53-mediated cell death [40].
Direct involvement of NF-κB in induction of cell death has been already reported in HEK293 cells upon at least 3 days of serum withdrawal [13]. The 1% serum treatment used in our study may have highlighted the greater sensitivity of FC1-depleted cells to undergoing apoptosis.
It has been demonstrated that the physical interaction between FC1 and PC2 prevents downregulation of PC2 [41] and that the loss of FC1 downregulates PC2 expression in vivo [27]. Moreover, it has been recently reported that the knockdown of TRPP2/polycystin-2 (PC2) channel increases both ER Ca2+ release and sensitivity to apoptosis in renal epithelial cells [28]. Therefore, FC1-depletion may cause PC2 reduction, leading to increased apoptosis. Interestingly, reduced PC2 levels were found to be associated to increased ATP-evoked Ca2+ levels in FC1-depleted cells (Fig. 7). The NF-κB-dependent apoptosis observed in these cells may therefore be supported by a Ca2+-dependent pathway [42] triggered by PC2 reduction, as highlighted in Fig. 8.

Schematics illustrating the contrasting roles of NF-κB in FC1-depleted and control kidney cells. Reduction in PC2 by FC1-depletion may increase ER Ca2+ release and mitochondrial Ca2+ signals, augmenting sensitivity to apoptosis [28]
Conclusions
Results presented herein show that loss of FC1 is one of several stimuli which induce NF-κB activation in renal tubular epithelial cells. Considering that NF-κB activity is involved in renal fibrotic events [25, 43–45], these findings provide evidence that NF-κB may be implicated in promoting or exacerbating the pathological conditions of ARPKD, a genetic disorder involving liver and kidney tissue and in which both proliferation, apoptosis and fibrosis are typical features. Moreover, the observation that apoptosis occurs in FC1-depleted cells in the absence of increased proliferation indicates that apoptosis may be a primary and proliferation-independent event in ARPKD. The discovery of an exaggerated NF-κB activation in PKHD1-deficient cells also suggests a new area for therapeutic investigation of this disease.
References
Guay-Woodford L (2003) Desmond R autosomal recessive polycystic kidney disease (ARPKD): the clinical experience in North America. Pediatrics 111:1072–1080
Zerres K, Rudnik-Schöneborn S, Senderek J, Eggermann T, Bergmann C (2003) Autosomal recessive polycystic kidney disease (ARPKD). J Nephrol 16:453–458
Menezes LF, Cai Y, Nagasawa Y et al (2004) Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int 66:1345–1355
Wang S, Zhang J, Nauli SM et al (2007) Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol 27:3241–3252
Harris PC, Torres VE (2009) Polycystic kidney disease. Annu Rev Med 60:321–337
Kuure S, Vuolteenaho R, Vainio S (2000) Kidney morphogenesis: cellular and molecular regulation. Mech Dev 92:31–45
Woo D (1995) Apoptosis and loss of renal tissue in polycystic kidney diseases. N Engl J Med 333:18–25
Kip SN, Hunter LW, Ren Q et al (2005) [Ca2+]i reduction increases cellular proliferation and apoptosis in vascular smooth muscle cells: relevance to the ADPKD phenotype. Circ Res 96:873–880
Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O (2006) Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 444:949–952
Starremans PG, Li X, Finnerty PE, Guo L et al (2008) A mouse model for polycystic kidney disease through a somatic in-frame deletion in the 5′ end of Pkd1. Kidney Int 73:1394–1405
Goilav B, Satlin LM, Wilson PD (2008) Pathways of apoptosis in human autosomal recessive and autosomal dominant polycystic kidney diseases. Pediatr Nephrol 23:1473–1482
Shen HM, Tergaonkar V (2009) NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis 14:348–363
Grimm S, Bauer MK, Baeuerle PA (1996) Schulze-Osthoff K.Bcl-2 down-regulates the activity of transcription factor NF-kappaB induced upon apoptosis. J Cell Biol 134:13–23
Baichwal VR, Baeuerle PA (1997) Activate NF-kappa B or die? Curr Biol 7:R94–R96
Burstein E, Duckett CS (2003) Dying for NF-kappaB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr Opin Cell Biol 15:732–737
Aguiari G, Trimi V, Bogo M et al (2008) Novel role for polycystin-1 in modulating cell proliferation through calcium oscillations in kidney cells. Cell Prolif 41:554–573
Wang S, Luo Y, Wilson PD, Witman GB, Zhou J (2004) The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. J Am Soc Nephrol 15:592–602
Nagano J, Kitamura K, Hujer KM et al (2005) Fibrocystin interacts with CAML, a protein involved in Ca2+ signaling. Biochem Biophys Res Commun 338:880–889
Aguiari G, Varani K, Bogo M et al (2009) Deficiency of polycystic kidney disease-1 gene (PKD1) expression increases A(3) adenosine receptors in human renal cells: Implications for cAMP-dependent signalling and proliferation of PKD1-mutated cystic cells. Biochim Biophys Acta 1792:531–540
Ward CJ, Yuan D, Masyuk TV et al (2003) Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet 12:2703–2710
Aguiari G, Banzi M, Gessi S et al (2004) Deficiency of polycystin-2 reduces Ca2+ channel activity and cell proliferation in ADPKD lymphoblastoid cells. FASEB J 18:884–886
Aguiari G, Campanella M, Manzati E et al (2003) Expression of polycystin-1 C-terminal fragment enhances the ATP-induced Ca2+ release in human kidney cells. Biochem Biophys Res Commun 301:657–664
Manzati E, Aguiari G, Banzi M et al (2005) The cytoplasmic C-terminus of polycystin-1 increases cell proliferation in kidney epithelial cells through serum-activated and Ca(2+)-dependent pathway(s). Exp Cell Res 304:391–406
Guijarro C, Egido J (2001) Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int 59:415–424
Zhang S, Lin ZN, Yang CF, Shi X, Ong CN, Shen HM (2004) Suppressed NF-kappaB and sustained JNK activation contribute to the sensitization effect of parthenolide to TNF-alpha-induced apoptosis in human cancer cells. Carcinogenesis 25:2191–2199
Borgatti M, Finotti A, Romanelli A et al (2004) Peptide nucleic acids (PNA)-DNA chimeras targeting transcription factors as a tool to modify gene expression. Curr Drug Targets 5:735–744
Kim I, Fu Y, Hui K et al (2008) Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J Am Soc Nephrol 19:455–468
Wegierski T, Steffl D, Kopp C et al (2009) TRPP2 channels regulate apoptosis through the Ca2+ concentration in the endoplasmic reticulum. EMBO J 28:490–499
Lanoix J, D’Agati V, Szabolcs M, Trudel M (1996) Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD). Oncogene 13:1153–1160
Boletta A, Qian F, Onuchic LF, Bhunia AK et al (2000) Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol Cell 6:1267–1273
Tao Y, Kim J, Faubel S et al (2005) Caspase inhibition reduces tubular apoptosis and proliferation and slows disease progression in polycystic kidney disease. Proc Natl Acad Sci USA 102:6954–6959
Woods JS, Ellis ME, Dieguez-Acuña FJ, Corral J (1999) Activation of NF-kappaB in normal rat kidney epithelial (NRK52E) cells is mediated via a redox-insensitive, calcium-dependent pathway. Toxicol Appl Pharmacol 154:219–227
Dieguez-Acuña FJ, Polk WW, Ellis ME, Simmonds PL, Kushleika JV, Woods JS (2004) Nuclear factor kappaB activity determines the sensitivity of kidney epithelial cells to apoptosis: implications for mercury-induced renal failure. Toxicol Sci 82:114–123
Edelstein CL (2005) What is the role of tubular epithelial cell apoptosis in polycystic kidney disease (PKD)? Cell Cycle 4:1550–1554
Torres VE, Harris PC (2006) Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol 2:40–55
Mai W, Chen D, Ding T et al (2005) Inhibition of Pkhd1 impairs tubulomorphogenesis of cultured IMCD cells. Mol Biol Cell 16:4398–4409
Cance WG, Golubovskaya VM (2008) Focal adhesion kinase versus p53: apoptosis or survival? Sci Signal 1(20):pe22
Yang J, Zhang S, Zhou Q et al (2007) PKHD1 gene silencing may cause cell abnormal proliferation through modulation of intracellular calcium in autosomal recessive polycystic kidney disease. J Biochem Mol Biol 40:467–474
Wu H, Lozano G (1994) NF-kappa B activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem 269:20067–20074
Ryan KM, Ernst MK, Rice NR, Vousden KH (2000) Role of NF-kappaB in p53-mediated programmed cell death. Nature 404:892–897
Kim I, Li C, Liang D et al (2008) Polycystin-2 expression is regulated by a PC2-binding domain in the intracellular portion of fibrocystin. J Biol Chem 283:31559–31566
Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R (2008) Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27:6407–6418
Morrissey J, Klahr S (1998) Transcription factor NF-kappaB regulation of renal fibrosis during ureteral obstruction. Semin Nephrol 18:603–611
Tamada S, Asai T, Kuwabara N et al (2006) Molecular mechanisms and therapeutic strategies of chronic renal injury: the role of nuclear factor kappaB activation in the development of renal fibrosis. J Pharmacol Sci 100:17–21
Guzik TJ (2007) Harrison DG.Endothelial NF-kappaB as a mediator of kidney damage: the missing link between systemic vascular and renal disease? Circ Res 101:227–229
Acknowledgments
We thank Dr. Johannes A. Schmid (Medical University, Vienna, Austria) for providing NFkB-GFP plasmid, Anna Forster for correcting the manuscript and I. Maestri and A. Canella for technical assistance. This work was supported by Cassa di Risparmio di Ferrara, Cassa di Risparmio di Cento, Italian MIUR COFIN 2008 and Regione Emilia Romagna (Ricerca Regione-Università) 2007–2009.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Mangolini, A., Bogo, M., Durante, C. et al. NF-κB activation is required for apoptosis in fibrocystin/polyductin-depleted kidney epithelial cells. Apoptosis 15, 94–104 (2010). https://doi.org/10.1007/s10495-009-0426-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-009-0426-7