
Abstract
We have previously shown that the Orf virus protein, ORFV125, is a potent inhibitor of the mitochondrial pathway of apoptosis and displays rudimentary sequence similarities to cellular anti-apoptotic Bcl-2 proteins. Here we investigate the proposal that ORFV125 acts in a Bcl-2-like manner to inhibit apoptosis. We show that the viral protein interacted with a range of BH3-only proteins (Bik, Puma, DP5, Noxa and all 3 isoforms of Bim) and neutralized their pro-apoptotic activity. In addition, ORFV125 bound to the active, but not the inactive, form of Bax, and reduced the formation of Bax dimers. Mutation of specific amino acids in ORFV125 that are conserved and functionally important in mammalian Bcl-2 family proteins led to loss of both binding and inhibitory functions. We conclude that ORFV125’s mechanism of action is Bcl-2-like and propose that the viral protein’s combined ability to bind to a range of BH3-only proteins as well as the active form of Bax provides significant protection against apoptosis. Furthermore, we demonstrate that the binding profile of ORFV125 is distinct to that of other poxviral Bcl-2-like proteins.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria are not only involved in the production of energy for a living cell, but also play a pivotal role in the process of apoptotic cell death. Mitochondrial apoptosis is strictly regulated by members of the Bcl-2 family. This protein family consists of one class of anti-apoptotic (Bcl-2-like) and two classes of pro-apoptotic (Bax-like and BH3-only) proteins that share 1–4 conserved Bcl-2 homology (BH) domains [1]. The Bax-like proteins, Bax and Bak are considered to be the executioners of the mitochondrial pathway of apoptosis. Upon activation, both proteins undergo considerable conformational change, form oligomeric structures and subsequently permeabilize the mitochondrial outer membrane. This results in a release of pro-apoptotic substances from the mitochondrial intermembrane space into the cytosol, which trigger caspase activation and apoptosis [2, 3]. The second class of pro-apoptotic molecules, the BH3-only proteins (Bim, Puma, Noxa, DP5/Hrk, Bad, Bmf, Bid and Bik) are the initiators of mitochondrial apoptosis. They are activated by a variety of cellular stresses and in turn directly or indirectly activate the Bax-like proteins.
The mechanism by which the anti-apoptotic Bcl-2 family members (Bcl-2, Bcl-w, Bcl-xL, Mcl-1 and A1) inhibit the action of their pro-apoptotic counterparts is still a matter of ongoing debate [4]. The indirect model proposes that anti-apoptotic Bcl-2-like proteins hold Bax and Bak in check, until the BH3-only proteins bind to them, thus preventing their anti-apoptotic function [5, 6]. In the direct model, the anti-apoptotic family members sequester the “activator” BH3-only proteins (Bim, tBid, and presumably Puma) that would otherwise directly activate Bax and Bak. The “sensitizer” BH3-only proteins (DP5/Hrk, Noxa, Bad, Bmf, Bik) cause a release of the “activator” proteins, by binding and inhibiting their anti-apoptotic counterparts [7, 8].
The BH1, BH2 and BH3 domains of the anti-apoptotic and Bax-like pro-apoptotic proteins form a prominent hydrophobic groove on the surface of these proteins that serves as a binding site for the α-helical BH3 domain of pro-apoptotic Bcl-2 family members [9–11]. Mutations of a conserved glycine and arginine in the BH1 domain or a tryptophan in the BH2 domain of anti-apoptotic Bcl-2 family members, such as Bcl-2 and Bcl-xL, prevented not only the interaction with the pro-apoptotic proteins, but also abolished their anti-apoptotic function [12, 13]. On the other hand, mutating a conserved leucine in the BH3 domain of pro-apoptotic members (Noxa, Bad, Bak and Bax) inhibited their interaction with anti-apoptotic members as well as their pro-apoptotic function [9, 14–16]. Interactions between Bcl-2 family members are thus crucial to their regulation and to their role in inhibition or promotion of apoptosis.
Viruses have developed a vast array of anti-apoptotic factors to overcome the innate response to viral infection, including proteins that mimic the anti-apoptotic Bcl-2 proteins. Viral Bcl-2 homologs have been found in large DNA viruses such as herpesviruses, adenoviruses, African swine fewer virus as well as poxviruses [17, 18]. There is only one recognized Bcl-2 homolog in the poxvirus family; FWPV039 of Fowlpox virus [19]. However, a number of anti-apoptotic proteins have been identified which do not show substantial sequence similarities to Bcl-2 proteins, but function in a Bcl-2-like manner to inhibit apoptosis. These include the F1L and N1L proteins of Vaccinia virus (Orthopoxvirus), and the M11L protein of Myxoma virus (Leporipoxvirus) [20–23].
We recently reported that Orf virus (ORFV), a member of the Parapoxvirus genus, encodes a unique anti-apoptotic protein, which shows little evidence of sequence similarities to other poxvirus proteins [24]. ORFV125 was shown to localize entirely to the mitochondria and to inhibit UV-induced apoptotic events downstream of this organelle, namely DNA fragmentation, caspase activation and cytochrome c release. An amino acid sequence alignment of ORFV125 with members of the Bcl-2 family revealed conserved residues of putative BH1 and BH3 domains as well as partial BH2 and BH4 domains. Furthermore, ORFV125 was able to entirely block UV-induced activation of the pro-apoptotic Bcl-2 family members Bak and Bax. We therefore hypothesized that ORFV125 functions in a Bcl-2-like manner to inhibit apoptosis.
In this study, we have addressed this hypothesis in two ways. We have determined the effect of mutating BH domain residues in ORFV125 that are conserved in and essential for the anti-apoptotic function of Bcl-2 family proteins. We have also investigated the ability of ORFV125 to interact with and inhibit apoptosis induced by pro-apoptotic Bcl-2 family members.
Materials and methods
Cell lines and culture conditions
143B cells were cultured in Eagle minimal essential medium (M0769, Sigma), while 293 EBNA, 293T and COS-7 cells were grown in Dulbecco’s modified Eagle medium (#058, GIBCO). All media were supplemented with 10% foetal bovine serum (PAA laboratories), 2 mM l-glutamine as well as a combination of 50 U/ml penicillin, 50 μg/ml streptomycin and 100 μg/ml kanamycin.
Expression vectors
For the construction of the pEGFPC1-Bcl-2G145E vector, the Bcl-2G145E coding region was amplified from pEF_Bcl-2G145EPGKpuro [25] (a gift from David Huang, WEHI, Australia) using the primers 5′BspEI-GGT CCG GAA TGG CGC ACG CTG GGA GAA CAG-3′ and 5′EcoR1-CCG AAT TCT CAC TTG TGG CCC AGA TAG GC-3′, and cloned into pEGFP-C1 to generate an EGFP-fusion protein.
To generate site-directed mutations of the codon region of ORFV125, the gene was amplified from the pEGFPC1-ORFV125wt construct as two separate PCR products using two wild-type primers binding at both ends of the gene and two mismatch primers binding to the to-be-mutated region (Table 1). The two PCR products overlapped, and together were used as templates for a third PCR, amplifying the entire coding region with the two wild-type primers. The PCR product was cloned into pEGFP-C1. To obtain FLAG-tagged versions of these proteins, the cDNAs were amplified from the pEGFPC1-constructs using 5′BglII-GGA GAT CTA TGG ACT ACA AGG ACG ACG ATG ACA AGA TGG CAA ACA GAG AAG AGA TTG-3′ and 5′XbaI-CCT CTA GAT TAT GTG CGC CGC AAC ACG C-3′, and were cloned into the BamHI/XbaI-digested pEF_Bcl-2PGKpuro [26] (a gift from David Huang).
The construction of pEGFPC1-ORFV125wt, -ts, -Bcl-2 as well as pEF_FLAG-ORFV125PGKpuro and -Bcl-2PGKpuro was described previously [24].
The set of plasmids (pEF_PGKhygro-derivatives) expressing the HA-tagged pro-apoptotic Bcl-2 family members, (hs) Bax, (hs) Bak, (hs) BimEL, (hs) BimL, (hs) BimS, (mm) Bid, (hs) tBid, (hs) Bik, (mm) Bmf, (mm) Bad, (mm) Noxa, (hs) Puma and (rn) DP5 [5, 6] was a kind gift from David Huang. The pEF_FLAG-(hs)Mcl-1PGKpuro [27] and pEF_FLAG-(hs)Bcl-xLPGKpuro [26] vectors were also supplied by David Huang.
Transfection
Cells were transfected with DNA using Lipofectin (Invitrogen) according to the manufacturer’s instructions. Medium was changed or topped up (for 293 EBNA and 293T cells) 6 h after transfection, and cells were incubated for various times at 37°C. In order to increase the transfection efficiency for the BH3-induced killing experiments, the cells were seeded and transfected at the same time. For this purpose, the Lipofectin/DNA mix was added directly to 500 μl of Opti-MEM containing 1 × 105 293T or COS-7 cells (24-wells) and incubated as described above.
Apoptosis induction
143B cells were UV-irradiated by inverting a drained 35-mm dish onto a transilluminator and exposing them for 4 s to 20 W/m2 (80 J/m2) UVC light (260 nm). Medium was then added and the cells were incubated for various times at 37°C.
Detection of active Bax and Bak
For the detection of active Bax and Bak, conformational-specific antibodies for Bax (1:250, 6A7, 556467, BD Biosciences Pharmingen) and Bak (1:150, Ab-1, AM03, Calbiochem) and an α-mouse Alexa Fluor 594 secondary antibody (1:400, A11020, Molecular Probes) were used in a staining protocol described previously [24].
DNA content assay
To detect DNA fragmentation, UV-treated 143B cells were permeabilized and stained with propidium iodide (PI) using a protocol described earlier [24]. Cells were analyzed for their resulting DNA content using a Becton–Dickinson FACScalibur. After exclusion of doublets and cell aggregates, the PI intensities of at least 2,000 single, PI- and GFP-positive cells were displayed in an FL2-area histogram. To ensure that any apparent functional differences between the EGFP-constructs did not result from differences in expression levels, the analysis was restricted to populations with similar EGFP-expression levels. The percentage of cells with reduced DNA content, which are located in the subG0/G1 peak (hypodiploid cells), was quantified using the CellQuest™ Pro analysis program from BD Bioscience (version 5.2).
Annexin-V staining
Floating and adherent cells (approximately 2 × 105 cells) were collected and resuspended in HEPES buffer (10 mM HEPES, 140 mM NaCl, 5 mM CaCl2, pH 7.4) supplemented with 2% bovine serum albumin (BSA). Cells were then resuspended in 100 μl HEPES buffer containing 2 μg/ml PI and 1 μl Annexin-V-biotin labeling reagent (1828690, Roche) and incubated for 15 min in the dark. Following a washing step in HEPES buffer containing 2% BSA, cells were resuspended in 100 μl HEPES buffer containing 4 ng/ml Streptavidin Cy5.5 antibody (S000-13, Rockland Immunochemicals, Inc.) and incubated for 20 min on ice in the dark. After a final washing step, cells were resuspended in HEPES buffer/2% BSA and were analyzed with a Becton and Dickinson FACScalibur (BD). The percentage of Annexin-positive, but PI-negative in at least 2,000 EGFP-positive cells with similar EGFP expression was calculated using the CellQuest Pro analysis program from BD (version 5.2).
Immunoprecipitation assays
Approximately 2–4 × 106 cells were incubated for 30 min at 4°C in lysis buffer (20 mM Tris–HCl; pH 7.4, 135 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol) supplemented with a protease inhibitor cocktail (complete mini EDTA-free, Roche) and 1% Triton X-100 or 1% Chaps. For immunoprecipitation of FLAG- or HA-tagged proteins, the soluble protein fractions were incubated with anti-HA- (A2095, Sigma) or anti-FLAG- (A2220, Sigma) conjugated agarose beads for 4 h at 4°C. After incubation, beads were washed 5× with TBS (10 mM Tris, 150 mM NaCl, pH 7.5), resuspended in SDS sample buffer (62.5 mM Tris–HCl; pH 6.8, 2% SDS, 10% glycerol, 0.01% bromphenol blue, 10% β-mercaptoethanol) and incubated at 95°C for 5 min. For immunoprecipitation of active Bax and Bak proteins, the soluble protein fractions were incubated with 2 μg active Bax (6A7) or 1 μg active Bak (Ab-1) antibody for 4 h at 4°C. After addition of protein G-conjugated agarose beads (E3403, Sigma), the samples were further incubated over night at 4°C. Beads were washed and resuspended in SDS sample buffer as described above. Cell lysates were prepared by resuspending the cell pellet in SDS sample buffer. FLAG- and HA-tagged proteins were detected by western blot using FLAG- (1:1,000, A8592, Sigma) and HA- (1:500-1:1,000, 2013819, Roche) horseradish-peroxidase-conjugated antibodies.
Protein cross-linking using 1,6-bismaleimidohexane (BMH)
293T cells were transfected with HA-Bax, HA-Bak and FLAG-ORFV125 expression plasmids using Lipofectin as described above. Six hours after transfection, medium was added containing z-VAD-fmk (Caspase Inhibitor I, 627610, Calbiochem) at a final concentration of 50 μM. Cells were harvested another 16 h later and resuspended in cross-link buffer (20 mM HEPES, 50 mM KCl, 2.5 mM MgCl2, 1 mM EDTA, 250 mM sucrose, pH 7.5) supplemented with a protease inhibitor cocktail (complete mini EDTA-free, Roche) at 1 × 107 cells/ml. BMH was added to a final concentration of 0.5 mM, and the cells were incubated at room temperature for 30 min. After centrifugation, the cell pellet was resuspended in SDS sample buffer (see above) and analyzed by western blot. The HA-tagged proteins were detected as described above. The detection of actin protein was performed as a loading control using a goat antibody (1:1,000, C-11, sc-1615, Santa Cruz Biotechnologies, Inc.) and an anti-goat horseradish-peroxidase-conjugated secondary antibody (1:10,000, A5420, Sigma).
Protein modelling of ORFV125
The structure-based alignment of mouse Bcl-xL (PDB: 1pq1, Bcl-xL:Bim peptide), human Mcl-1 (PDB: 2pqk, Mcl-1:Bim peptide) and human Bcl-w (PDB: 1o0l, Bcl-w) was performed using Mustang (v.3) [28]. The ORFV125 sequence was added to this alignment according to the sequence-based alignment presented in earlier studies [24]. Using the structural alignment, 3D models of ORFV125, with and without a Bim BH3 peptide incorporated, were generated with the Modeller software 9v3 (copyright © 1989–2008 Andrej Sali, http://salilab.org/modeller/modeller.html). The same PDB files were used for the generation of both ORFV125 models, however, the Bim peptide coordinates from 1pq1 and 2pqk were excluded when building the model of ORFV125 without the peptide. For each model, five protein models of ORFV125 were generated and evaluated, and the model with the lowest objective function was chosen. All structures and models were analyzed using Pymol 1.0 (http://pymol.sourceforge).
Statistical analysis
A one-way ANOVA with a Tukey–Kramer multiple comparison post test (P < 0.05) was performed using GraphPad InStat version 3.0a for Macintosh (www.graphpad.com).
Results
ORFV125 shares predicted structural features with Bcl-2 proteins
Standard BLAST analyses failed to detect significant sequence similarities between ORFV125 and cellular anti-apoptotic Bcl-2 family members. However, a Clustal W alignment of these proteins revealed an overall sequence identity of 5–17% and showed that the viral protein possesses recognizable BH1 and BH3 domains as well as partial BH2 and BH4 domains [24]. In addition, the HHpred program [29], which detects protein homology on the basis of secondary structure similarities, identified the cellular Bcl-2 family members as the four most similar proteins to ORFV125, with the top-ranked protein, Bcl-w, displaying a P-value of 5E-4.
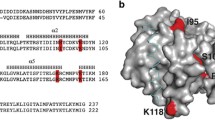
To further investigate the Bcl-2-like features of ORFV125, a structural alignment and 3D model of the viral protein were created using the structures of three anti-apoptotic Bcl-2 proteins, human Mcl-1, mouse Bcl-xL and human Bcl-w as templates (Fig. 1a, b). The tertiary structure of cellular Bcl-2 family members is, apart from the primarily unstructured BH3-only proteins [30], well conserved. The proteins consist of one or two predominantly hydrophobic α-helices, which are surrounded by six/seven amphipathic α-helices [31]. The modeled ORFV125 structure resembled the Bcl-w protein more closely than Bcl-xL and Mcl-1, possessing 7 α-helices in similar positions and orientation as 7 of the 8 α-helices in Bcl-w (Fig. 1b). The helices in the ORFV125 model are arranged as one hydrophobic central α-helix (α5) surrounded by 6 amphipathic helices; α1, α2, α6 and α7 on one side and α3 and α4 on the other side (Fig. 1b). A kink in α6 (D129) causes a change in direction and leads directly into α7. This helix is, however, shorter in the viral protein’s structure than in Bcl-w. This ORFV125 model does not contain a helix equivalent to α8. However, the loop region in this area exhibits a helical fold, and other models generated in Modeller exhibited an α-helix at position A136-G140 (data not shown).

ORFV125 is predicted to have a Bcl-2-like structure. a Structure-based Mustang alignment of 3 anti-apoptotic Bcl-2 family members [mouse Bcl-xL (PDB: 1pq1), human Mcl-1 (PDB: 2pqk) and human Bcl-w (PDB: 1o0l)] with the ORFV125 sequence incorporated. The BH domains of Bcl-xL are indicated as colored boxes. Helix positions within the Bcl-2 proteins and the predicted helix positions of the ORFV125 model are highlighted in gray within the alignment. Amino acids that were not used for the generation of the ORFV125 model are colored in blue. The residue numbers below the alignment refer to the ORFV125 sequence. Red asterisks indicate amino acids in ORFV125 chosen for mutation, as these residues are functionally conserved in cellular Bcl-2 family members. b Cartoon depiction of the ORFV125 model (left panel) and an overlay with the Bcl-w structure (right panel). Helices are numbered as shown in a. c Comparing the known interactions of the mouse Bim peptide and mouse Bcl-xL with those predicted for Bim and ORFV125. The sequence of the BH3 peptide of Bim is positioned in the middle in blue, while the interacting residues of Bcl-xL and ORFV125 are boxed above and below the Bim sequence. Within the boxes, Bcl-xL amino acids are located before the dash and the corresponding residues of ORFV125 (from a) are after the dash. Identical amino acids and conserved substitutions are colored in black, while non-identical residues are red. A question mark indicates that there is no corresponding residue in ORFV125 at that position
The model shows that the predicted BH domains are in close proximity in the viral protein, forming a potential hydrophobic groove on the protein’s surface, as occurs in the cellular Bcl-2 proteins. To identify the residues in the groove that might allow binding of pro-apoptotic BH3 domains, the human Mcl-1 and mouse Bcl-xL structures, in complex with a Bim peptide, were used as templates to build a second model of ORFV125 in which the Bim BH3 domain was incorporated. Most of the residues in the ORFV125 groove that are predicted to contact the Bim peptide are conserved with respect to the contacting amino acids in Bcl-xL (Fig. 1c, black amino acids). Hydrophobic amino acids in ORFV125 form predicted hydrophobic pockets, which are able to accommodate the hydrophobic residues of the Bim peptide, and polar residues of ORFV125 make predicted contacts to polar residues in Bim. In addition, ORFV125 shares residues with Bcl-xL, as well as other Bcl-2 family members, that have been shown to be important for the function of these proteins. These include a leucine (L40) in the BH3 domain, a glycine (G86) and an arginine (R87) in the BH1 domain and a tryptophan (W133) in the BH2 domain (Fig. 1a, red asterisks).
Mutation of conserved BH domain residues in ORFV125 causes loss of anti-apoptotic function
We had previously shown that ORFV125 inhibits UV-induced DNA fragmentation and Bax/Bak activation [24]. To determine if the conserved residues in the BH1-, BH2- and BH3-like domains of ORFV125 play a role in the viral protein’s anti-apoptotic function, mutated ORFV125 proteins fused to EGFP were created and tested for their ability to inhibit UV-induced DNA fragmentation. While around 30% of cells transfected with a negative control EGFP-fusion protein containing only the mitochondrial-targeting sequence of ORFV125 (ORFV125ts) underwent apoptosis, only 8% of the ORFV125wt-transfected cells were apoptotic (Fig. 2a). Mutation of the leucine of the BH3 domain (ORFV125L40A), glycine or arginine of the BH1 (ORFV125G86A and -R87A) or the tryptophan of the BH2 domain (ORFV125W133A) caused the viral protein to lose its ability to inhibit UV-induced DNA fragmentation. This loss was equivalent to that seen with a corresponding Bcl-2 BH1 domain mutant (Bcl-2G145E) (Fig. 2a). Consistent with this loss of ability to inhibit DNA fragmentation, it was found that each of these mutations also resulted in ORFV125 and Bcl-2 losing the ability to inhibit UV-induced Bax or Bak activation (Fig. 2b, c).

Mutation of conserved residues in the ORFV125 BH domains causes loss of anti-apoptotic function. 143B cells were transfected with constructs expressing EGFP fusions of wild-type ORFV125 (wt), the mitochondrial targeting sequence of ORFV125 (ts), ORFV125 mutated proteins (L40A, G86A, R87A, W133A), wild-type Bcl-2 (wt) or the Bcl-2 BH1 domain mutant (G145E). Transfected cells were UV irradiated 30 h (a) or 20 h (b, c) later. a The DNA content of permeabilized, PI-stained cells was assessed 17 h after UV irradiation as described in “Materials and methods”, and the percentage of hypodiploid (apoptotic) cells in each population was calculated. The data presented are averages of three independent experiments with SD indicated (+ P < 0.05, # P < 0.01 and * P < 0.001 compared with UV-irradiated ORFV125wt; ^ P < 0.001 compared with UV-treated Bcl-2wt). b, c Bax and Bak activation was assessed 6 h after UV irradiation as described in “Materials and methods”, and the percentage of EGFP-positive cells with active Bax (b) or Bak (c) in 4 different microscope fields was calculated and averaged. The results presented are an average of 2 (for +UV samples, range indicated) or one independent experiment (for −UV samples)
These data show that ORFV125 shares key functional amino acids with the Bcl-2 family members and that these are also important for the anti-apoptotic function of the viral protein.
ORFV125 interacts with a subset of BH3-only proteins
We next wanted to determine if ORFV125’s ability to block UV-induced activation of Bax and Bak (Fig. 2b, c) is mediated by direct interactions with these proteins or by it blocking the activity of BH3-only family members upstream of Bax and Bak activation. To address this, the ability of wild-type and mutated ORFV125 proteins to interact with pro-apoptotic Bcl-2 family members was tested in co-immunoprecipitation assays. Since highly specific antibodies are not available for all of these proteins, and to facilitate comparison of ORFV125’s interaction with each of them, we used exogenous expression of HA-tagged pro-apoptotic proteins in conjunction with protocols that have been used successfully to view interactions of cellular Bcl-2 family members [5, 32]. We found that ORFV125 co-precipitated with the BH3-only proteins Bik, Puma and the 3 isoforms of Bim (BimEL, BimL, BimS), and showed weaker binding to Noxa and DP5 (Fig. 3a). We did not detect interactions between the viral protein and Bad, Bmf, Bid or tBid (Fig. 3a), even with extended exposure times (data not shown), nor were there detectable interactions with the Bax-like proteins, Bax and Bak (Fig. 3a). Under equivalent conditions, we observed the expected interactions between the cellular anti-apoptotic Bcl-2 family members, Bcl-2, Bcl-xL and Mcl-1 and the pro-apoptotic proteins Bax, Bak, Bmf, Bad, Bid and tBid (supplemental Fig. 1), indicating that the observed lack of interaction with ORFV125 was not due to technical limitations.

The ability of wild-type and mutated ORFV125 proteins to bind to pro-apoptotic Bcl-2 proteins. a ORFV125 interacts with a subset of pro-apoptotic Bcl-2 family members. 293 EBNA cells were co-transfected with vectors expressing FLAG-ORFV125 wild-type and the indicated HA-tagged pro-apoptotic Bcl-2 family members. Twenty-four hours after transfection, 2 × 106 cells were harvested for immunoprecipitation (IP) with FLAG (FL) and HA beads in 1% Triton X-100 buffer. Precipitated and co-precipitated proteins from 1 × 106 cells were then detected by western blot (WB) using α-FLAG (α-FL) or α-HA antibodies. b Mutation in the ORFV125 BH domains alters the ability of ORFV125 to bind to Puma, BimS and Bik. 293 EBNA cells were co-transfected with vectors expressing FLAG-ORFV125wt (Wt), -L40A (L), -G86A (G), -R87A (R) or -W133A (W) in combination with HA-tagged Puma, BimS and Bik. Twenty-four hours after transfection, cells were harvested for immunoprecipitation and western blot as described in (a)
Next, the ability of the mutated ORFV125 proteins to bind to Puma, BimS or Bik was investigated. Interestingly, the ability of these proteins to interact with the pro-apoptotic Bcl-2 family members differed considerably (Fig. 3b). We did not detect interaction between the L40A or R87A mutated proteins and Puma, BimS or Bik (Fig. 3b), even with exposure times of up to 3 h (data not shown). On the other hand, the G86A protein bound weakly to Puma and Bik, but did not interact with BimS, while W133A retained its full binding ability for Bik, but interacted only weakly with BimS and Puma.
Together, these results indicate that ORFV125 acts in a Bcl-2-like manner to inhibit apoptosis; it interacts with a subset of pro-apoptotic Bcl-2 proteins, and its conserved BH domain residues are necessary for these interactions. However, the viral protein is not identical to the cellular anti-apoptotic Bcl-2 proteins, as it does not directly bind to Bax and Bak.
ORFV125 inhibits apoptosis induced by pro-apoptotic proteins
Our analysis of ORFV125’s binding profile suggested that ORFV125 inhibits apoptosis by binding to BH3-only proteins, but not to Bax or Bak. To test this, we investigated whether ORFV125 could prevent cell death induced by over-expression of both classes of pro-apoptotic Bcl-2 family members. Over-expression of Bax and Bak results in their spontaneous activation in a manner that is independent of BH3-only proteins, thus providing a means of studying ORFV125’s effect on the Bax-like proteins directly, without the influence of the BH3-only proteins.
We first showed that transient transfection of either Bax-like (Bax or Bak) or BH3-only proteins (BimS, Bik, tBid, DP5 or Puma) induced apoptosis in 293T or COS-7 cells (Fig. 4). We then tested apoptosis induction when wild-type or mutated ORFV125 proteins were co-expressed with each of these pro-apoptotic proteins (Fig. 4). The preceding immunoprecipitation experiments had shown that the HA-tagged pro-apoptotic proteins are adequately expressed in the presence of ORFV125 (Fig. 3). Co-transfection of ORFV125 strongly inhibited apoptosis induction by BimS and Bik, had an intermediate inhibitory effect on Puma, weakly inhibited DP5-induced apoptosis and had little effect on tBid (Fig. 4a, b). ORFV125 did not significantly inhibit Bak-induced apoptosis, although similar to the result seen with tBid, there was a slight reduction in apoptosis at the highest concentration of ORFV125 (Fig. 4a). Interestingly, it did show significant inhibition of apoptosis induced by over-expression of Bax (Fig. 4a). Neither the ORFV125L40A nor the -R87A proteins inhibited cell death induced by BimS, Bik or Puma (Fig. 4c, d). On the other hand, the ORFV125G86A and -W133A proteins retained some activity, as they fully inhibited BimS-induced apoptosis and had a partial influence on Bik.

The ability of ORFV125 wild-type and mutated proteins to inhibit apoptosis induced by pro-apoptotic Bcl-2 family members. a, b ORFV125 inhibits apoptosis induced by specific pro-apoptotic Bcl-2 family members. 293T (a) or COS-7 (b) cells were transfected with 0.4 μg of vector expressing FLAG (vector-only), Bax, Bak, BimS, Bik or Puma or 0.8 μg of tBid or DP5, and co-transfected with increasing amounts of the EGFP-ORFV125wt expression vector. These amounts of the pro-apoptotic constructs had previously been optimized to obtain an apoptosis rate of 20–30%. The total amount of EGFP protein in each transfection was kept consistent by adding up to 0.4 μg of the negative control construct expressing EGFP-ORFV125ts. Eighteen hours (a) or 22 h (b) after transfection, cells were stained with Annexin-V and PI and analyzed by flow cytometry. The percentage of EGFP-transfected cells that were Annexin positive, but PI negative, was calculated. Data are presented as the average of 3 independent experiments with SD indicated (+ P < 0.05, # P < 0.01 and * P < 0.001 compared with 0 μg EGFP-ORFV125wt). c, d Mutation in the ORFV125 BH domains alters the ability of ORFV125 to inhibit Puma-, BimS- and Bik-induced apoptosis. 293T (c) and COS-7 (d) cells were transfected with 0.4 μg of vector expressing FLAG (vector-only), BimS, Bik or Puma, and co-transfected with 0.2 μg (c) or 0.4 μg (d) of constructs expressing EGFP-ORFV125wt, -ts, -L40A, -G86A, -R87A or -W133A. Eighteen hours (c) or 22 h (d) after transfection, cells were stained with Annexin-V and PI and analyzed by flow cytometry as described in a, b. Data are presented as the average of 3 independent experiments with SD indicated (+ P < 0.05, # P < 0.01 and * P < 0.001 compared with EGFP-ORFV125wt)
Taken together, this shows that ORFV125 prevents apoptosis induced by various BH3-only proteins, and also exerts an anti-apoptotic effect on Bax.
ORFV125 binds the active form of Bax and reduces Bax dimerization
We had shown that ORFV125 did not co-precipitate with either Bax or Bak when they were co-expressed in 293 EBNA cells (Fig. 3a) and had little effect on apoptosis induced by over-expression of Bak, but did inhibit Bax-induced apoptosis in 293T cells (Fig. 4a). Prompted by this apparent discrepancy between an ability to inhibit Bax-induced apoptosis while failing to co-precipitate with Bax, we further investigated the effect of ORFV125 on over-expressed Bax and Bak in 293T cells (Fig. 5). Following transient transfection of Bax or Bak into these cells, the activated forms of both proteins were readily detected using conformational-specific antibodies, which recognize N-terminal epitopes that are exposed upon activation of these proteins (Fig. 5a, b). The presence or absence of ORFV125 did not inhibit the conformational change indicating that the viral protein did not prevent spontaneous activation of exogenously expressed Bax or Bak. When we specifically immunoprecipitated the active form of Bax or Bak from lysates of these cells, the viral protein co-precipitated with active Bax, but not with active Bak (Fig. 5c, left panel). These results mirrored the ability of ORFV125 to inhibit apoptosis induced by Bax while having little effect on Bak-induced apoptosis (Fig. 4a). The results also suggested that the lack of a detectable interaction between ORFV125 and Bax in 293 EBNA cells could be due to lack of activation of the exogenously expressed protein in these cells. Indeed, very little active Bax and a significantly reduced amount of active Bak was immunoprecipitated from the 293 EBNA cells (Fig. 5c, right panel) compared with 293T cells.

ORFV125 binds to active Bax and inhibits Bax dimerization. a, b ORFV125 does not inhibit activation of Bax or Bak. 293T cells were co-transfected with 0.4 μg of vector expressing HA-Bax or HA-Bak and 0.4 μg of vector expressing EGFP-ORFV125wt or -ORFV125ts. Eighteen hours later, cells were stained with the α-Bax (6A7) (a), α-Bak (Ab-1) (b) conformational-specific antibodies that can detect activated forms of these 2 proteins, and examined by fluorescence microscopy. The percentage of EGFP-positive cells with active Bax or Bak in 4 different microscope fields was calculated and averaged. The results presented are the average of 3 independent experiments with SD indicated (# P < 0.01 and * P < 0.001 compared with the EGFP-ORFV125ts-only control). c ORFV125 binds to active Bax, but not active Bak. 293T or 293 EBNA cells were transfected with a vector expressing FLAG-ORFV125 and co-transfected with HA-Bax or -Bak constructs. Twenty-two hours after transfection, active Bax or Bak were immunoprecipitated (IP) from 4 × 106 cells with α-Bax (6A7) and α-Bak (Ab-1) conformational-specific antibodies in 1% Chaps buffer. 2 × 106 cells were used for detection of precipitated and co-precipitated proteins by western blot (WB) using α-FLAG (α-FL) or α-HA antibodies. An aliquot of the cell lysate (2 × 105 cells) was loaded to confirm expression of each protein. d ORFV125 inhibits Bax, but not Bak dimerization. 293T cells were transfected with the FLAG-ORFV125 expression vector and co-transfected with a vector expressing HA-Bax or -Bak. Twenty-two hours after transfection, samples were cross-linked with BMH and analyzed by western blot using α-HA antibody. Actin served as a loading control
We next investigated whether ORFV125 prevents the oligomerization of Bax or Bak that occurs subsequent to the conformational change. 293T cells were transfected with Bax or Bak in the presence or absence of ORFV125, and 22 h later cross-linking was performed using BMH. Bax dimers were readily detected in cells transfected with Bax but were substantially reduced in cells co-transfected with ORFV125, along with a corresponding increase in the amount of Bax monomer (Fig. 5d, left panel). In contrast, ORFV125 did not inhibit Bak dimerization (Fig. 5d, right panel).
Taken together, this suggests that ORFV125 binds to the active form of Bax and diminishes its ability to form dimers, but does not interact with active Bak or inhibit Bak dimerization.
Discussion
The expression of viral factors that mimic functions of cellular proteins and inhibit host responses to viral infection is a common theme amongst large DNA viruses. Many such viruses encode homologs of Bcl-2 proteins that counteract apoptosis induction. ORFV, a member of the poxvirus family, was found to express a protein, ORFV125, which shows rudimentary primary sequence similarities to members of the Bcl-2 proteins [24]. This study set out to test the hypothesis that ORFV125 functions in a Bcl-2-like manner to inhibit apoptosis.
We were able to show that functionally important residues in the BH domains of cellular Bcl-2 proteins are conserved in ORFV125, and mutation of these residues abrogates the anti-apoptotic activity of the viral protein. Furthermore, ORFV125 binds to and neutralizes the activity of pro-apoptotic Bcl-2 proteins. These results clearly define ORFV125 as a viral Bcl-2-like protein.
Our results revealed that ORFV125 binds a set of BH3-only proteins, and this binding correlates strongly with ORFV125’s ability to inhibit apoptosis induced by those proteins (Table 2, Part A). This suggests that ORFV125’s interaction with this subset of BH3-only proteins may prevent them from triggering apoptosis. The viral protein displays a range of anti-apoptotic potencies. While it can interact with and inhibit BimS and Bik very strongly, its binding and inhibitory effect on Puma and DP5 is moderate to weak. In contrast, ORFV125 does not interact with Bmf, Bad, Bid and tBid, and has little if any ability to prevent apoptosis induced by tBid. The question remains as to why ORFV125 does not bind to all BH3-only proteins. It is possible that the viral protein, as it is entirely localized to the mitochondria, only interacts with BH3-only proteins that insert into the mitochondrial membrane via a membrane-targeting motif. Or it may only bind to proteins that can induce apoptosis by themselves, or to one member of a set of proteins, such as Bad and Noxa, that would only trigger apoptosis in combination [5, 6]. Another possibility is that ORFV125 only targets BH3-only proteins that are activated in response to ORFV infection. It is not known which pro-apoptotic pathways are activated following ORFV infection, but studies with other viruses have shown activation of Bik, Bim, Noxa and Bid [33–35]. ORFV125 binds to each of these BH3-only proteins, apart from Bid, suggesting that they might be involved in the response to ORFV. ORFV125’s inability to bind to full-length Bid or to significantly inhibit apoptosis induced by tBid, suggests that the viral protein may not be able to prevent apoptosis triggered by the death-receptor signaling pathway via caspase-8-mediated cleavage of Bid. However, ORFV has been shown to express a viral IL-10 [36], which may be involved in the downregulation of tumor necrosis factor-α, and may therefore have an indirect influence on the production of tBid [37]. In future studies it will be interesting to investigate which apoptotic pathways are induced by ORFV infection, to identify the particular Bcl-2 family members involved in this process and determine if the interactions described in this study also occur during virus infection.
In addition to ORFV125’s ability to bind to BH3-only proteins, we found that the viral protein can also interact with the active form of Bax and substantially reduces the formation of Bax dimers, providing additional protection against apoptosis. This latter observation is reminiscent of the adenoviral Bcl-2 homolog, E1B19K, which has been shown to interact with Bax only after a conformational change induced by tBid, and to block Bax oligomerization [38]. Although ORFV125 may have some ability to inhibit Bak-induced apoptosis following over-expression (Fig. 4a), the viral protein is not able to bind directly to active Bak or inhibit its dimerization (Fig. 5c, d), suggesting that ORFV125’s anti-apoptotic activity is not directed at this pro-apoptotic protein. Interestingly, the selective ability of the viral protein to act against Bax, but not Bak is similar to that of the cellular Bcl-2 protein, Bcl-b [39].
The strong correlation between the binding and inhibitory abilities of ORFV125 favors the idea of a direct action against the BH3-only proteins, although we cannot completely rule out the possibility that ORFV125 may inhibit BH3-only-induced apoptosis by binding to active Bax rather than the BH3-only protein itself (Fig. 4). However, our results indicate that ORFV125’s ability to entirely block UV-induced activation of Bax and Bak (Fig. 2b, c and [24]), is most likely based on its sole interaction and inhibition of activated BH3-only proteins, as ORFV125 has no influence on the spontaneous, BH3-only-independent activation of over-expressed Bax and Bak (Fig. 5a, b). We therefore propose that ORFV125 provides significant protection against apoptosis by its combined ability to bind to both BH3-only proteins and to the activated form of Bax (Fig. 6).

ORFV125 provides two levels of protection against apoptosis. In response to a death signal (e.g. UV irradiation), BH3-only proteins are activated and trigger Bax and Bak to undergo conformational change (Bax* and Bak*), leading to their oligomerization. ORFV125 interacts with a range of BH3-only proteins, thus preventing them from activating Bax and Bak. In addition, ORFV125 can inhibit the apoptosis-inducing activity of Bax by directly binding to its activated form and thereby preventing its oligomerization
Mutation of residues of ORFV125 that are conserved and functionally important in cellular Bcl-2 proteins resulted in a complete loss of the viral protein’s ability to inhibit UV-induced DNA fragmentation and activation of Bax and Bak. However, some of these mutated proteins remained able to bind and inhibit some individual BH3-only proteins (Table 2, Part B), abilities that were obviously insufficient to block the effects of UV, a broad apoptosis inducer that is likely to activate multiple BH3-only proteins [40]. Overall, there was a good correlation between the binding and inhibitory abilities of the mutated proteins. There were a small number of exceptions, such as G86A’s interactions with BimS and W133A’s with Bik, indicating that other factors may play a role. The results seen with the L40A mutation were somewhat puzzling at first, since in cellular Bcl-2 proteins this residue has been primarily linked to pro-apoptotic rather than anti-apoptotic functions [9, 14–16]. However, a similar result has been reported for the equivalent Bcl-2 mutated protein, L97A, which no longer binds to full-length Bad [15]. 3D modelling of ORFV125 showed that the L40 residue is located in the hydrophobic core of the protein (data not shown), and mutation of this residue might therefore lead to an altered protein fold, abrogating the interaction with BH3-peptides as well as the anti-apoptotic function. The results of mutational analyses provide strong evidence of the functional significance of the sequence/secondary structure similarities between ORFV125 and Bcl-2 family members we previously reported [24].
The Bcl-2-like properties of ORFV125 place it in a growing cluster of poxviral Bcl-2-like factors that includes F1L, N1L, M11L and FWPV039. Although not initially regarded as Bcl-2 homologs, the presence of characteristic residues of BH domains [24] and the structural demonstrations of a Bcl-2-like fold (Fig. 1 and [21, 22, 41–43]) leave no doubt as to the Bcl-2-like nature of these viral proteins. Furthermore, functional analyses showed that all proteins inhibit the mitochondrial pathway of apoptosis [20, 22, 23, 44] and bind to pro-apoptotic Bcl-2 family members (Table 3), defining the poxviral proteins as a group of functional homologs of the Bcl-2 family.
Although the full set of comparative data is not available, it appears that the binding profile of ORFV125 differs from that of other poxviral Bcl-2-like proteins (Table 3). While the latter proteins have been found to bind to some BH3-only proteins, their main target seems to be the Bax-like proteins. All four proteins have been shown to interact constitutively with Bak, and recent evidence suggests that they can also bind to at least the activated form of Bax [21, 22, 42–48]. ORFV125’s failure to interact with Bak combined with its ability to bind a wide range of BH3-only proteins therefore clearly distinguishes the ORFV protein from its poxvirus relatives.
Interestingly, while poxviruses commonly encode multiple inhibitors of apoptosis that interfere with the mitochondrial as well as death receptor pathway or inhibit caspases [49], we have not identified any additional anti-apoptotic proteins encoded by ORFV. Nor have we been able to create a stable ORFV125-deletion virus, as might be the case if it were the only ORFV apoptosis inhibitor. It is therefore possible, that the broad BH3-only binding profile displayed by ORFV125 is linked to the absence of additional ORFV anti-apoptotic factors.
Abbreviations
- Bcl-2:
-
B-cell leukemia/lymphoma 2
- BH:
-
Bcl-2 homology
- ORFV:
-
Orf virus
References
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59
Griffiths GJ, Dubrez L, Morgan CP et al (1999) Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J Cell Biol 144:903–914
Er E, Oliver L, Cartron PF, Juin P, Manon S, Vallette FM (2006) Mitochondria as the target of the pro-apoptotic protein Bax. Biochim Biophys Acta 1757:1301–1311
Adams JM, Cory S (2007) Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol 19:488–496
Chen L, Willis SN, Wei A et al (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17:393–403
Willis SN, Fletcher JI, Kaufmann T et al (2007) Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315:856–859
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2:183–192
Kim H, Rafiuddin-Shah M, Tu HC et al (2006) Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 8:1348–1358
Sattler M, Liang H, Nettesheim D et al (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275:983–986
Denisov AY, Chen G, Sprules T, Moldoveanu T, Beauparlant P, Gehring K (2006) Structural model of the BCL-w-BID peptide complex and its interactions with phospholipid micelles. Biochemistry 45:2250–2256
Czabotar PE, Lee EF, van Delft MF et al (2007) Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci USA 104:6217–6222
Yin XM, Oltvai ZN, Korsmeyer SJ (1994) BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 369:321–323
Cheng EH, Levine B, Boise LH, Thompson CB, Hardwick JM (1996) Bax-independent inhibition of apoptosis by Bcl-XL. Nature 379:554–556
Oda E, Ohki R, Murasawa H et al (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053–1058
Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ (1997) BH3 domain of BAD is required for heterodimerization with BCL-XL and pro-apoptotic activity. J Biol Chem 272:24101–24104
Wang K, Gross A, Waksman G, Korsmeyer SJ (1998) Mutagenesis of the BH3 domain of BAX identifies residues critical for dimerization and killing. Mol Cell Biol 18:6083–6089
Cuconati A, White E (2002) Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev 16:2465–2478
Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G (2008) Viral control of mitochondrial apoptosis. PLoS Pathog 4:e1000018
Afonso CL, Tulman ER, Lu Z, Zsak L, Kutish GF, Rock DL (2000) The genome of fowlpox virus. J Virol 74:3815–3831
Wasilenko ST, Stewart TL, Meyers AF, Barry M (2003) Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc Natl Acad Sci USA 100:14345–14350
Aoyagi M, Zhai D, Jin C et al (2007) Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci 16:118–124
Cooray S, Bahar MW, Abrescia NG et al (2007) Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J Gen Virol 88:1656–1666
Everett H, Barry M, Lee SF et al (2000) M11L: a novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. J Exp Med 191:1487–1498
Westphal D, Ledgerwood EC, Hibma MH, Fleming SB, Whelan EM, Mercer AA (2007) A novel Bcl-2-like inhibitor of apoptosis is encoded by the parapoxvirus ORF virus. J Virol 81:7178–7188
O’Reilly LA, Huang DC, Strasser A (1996) The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J 15:6979–6990
Huang DC, Cory S, Strasser A (1997) Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene 14:405–414
Moriishi K, Huang DC, Cory S, Adams JM (1999) Bcl-2 family members do not inhibit apoptosis by binding the caspase activator Apaf-1. Proc Natl Acad Sci USA 96:9683–9688
Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM (2006) MUSTANG: a multiple structural alignment algorithm. Proteins 64:559–574
Soding J (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951–960
Hinds MG, Smits C, Fredericks-Short R et al (2007) Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ 14:128–136
Petros AM, Olejniczak ET, Fesik SW (2004) Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta 1644:83–94
Willis SN, Chen L, Dewson G et al (2005) Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19:1294–1305
Subramanian T, Vijayalingam S, Lomonosova E, Zhao LJ, Chinnadurai G (2007) Evidence for involvement of BH3-only proapoptotic members in adenovirus-induced apoptosis. J Virol 81:10486–10495
Sun Y, Leaman DW (2005) Involvement of Noxa in cellular apoptotic responses to interferon, double-stranded RNA, and virus infection. J Biol Chem 280:15561–15568
Belov GA, Romanova LI, Tolskaya EA, Kolesnikova MS, Lazebnik YA, Agol VI (2003) The major apoptotic pathway activated and suppressed by poliovirus. J Virol 77:45–56
Fleming SB, McCaughan CA, Andrews AE, Nash AD, Mercer AA (1997) A homolog of interleukin-10 is encoded by the poxvirus orf virus. J Virol 71:4857–4861
Becherel PA, LeGoff L, Frances C et al (1997) Induction of IL-10 synthesis by human keratinocytes through CD23 ligation: a cyclic adenosine 3′,5′-monophosphate-dependent mechanism. J Immunol 159:5761–5765
Perez D, White E (2000) TNF-alpha signals apoptosis through a bid-dependent conformational change in Bax that is inhibited by E1B 19K. Mol Cell 6:53–63
Zhai D, Jin C, Huang Z, Satterthwait AC, Reed JC (2008) Differential regulation of Bax and Bak by anti-apoptotic Bcl-2-family proteins, Bcl-B and Mcl-1. J Biol Chem 283:9580–9586
Kulms D, Zeise E, Poppelmann B, Schwarz T (2002) DNA damage, death receptor activation and reactive oxygen species contribute to ultraviolet radiation-induced apoptosis in an essential and independent way. Oncogene 21:5844–5851
Douglas AE, Corbett KD, Berger JM, McFadden G, Handel TM (2007) Structure of M11L: a myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2. Protein Sci 16:695–703
Kvansakul M, van Delft MF, Lee EF et al (2007) A structural viral mimic of prosurvival Bcl-2: a pivotal role for sequestering proapoptotic Bax and Bak. Mol Cell 25:933–942
Kvansakul M, Yang H, Fairlie WD et al (2008) Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ 15:1564–1571
Banadyga L, Gerig J, Stewart T, Barry M (2007) Fowlpox virus encodes a Bcl-2 homologue that protects cells from apoptotic death through interaction with the proapoptotic protein Bak. J Virol 81:11032–11045
Wang G, Barrett JW, Nazarian SH et al (2004) Myxoma virus M11L prevents apoptosis through constitutive interaction with Bak. J Virol 78:7097–7111
Su J, Wang G, Barrett JW, Irvine TS, Gao X, McFadden G (2006) Myxoma virus M11L blocks apoptosis through inhibition of conformational activation of Bax at the mitochondria. J Virol 80:1140–1151
Wasilenko ST, Banadyga L, Bond D, Barry M (2005) The vaccinia virus F1L protein interacts with the proapoptotic protein Bak and inhibits Bak activation. J Virol 79:14031–14043
Banadyga L, Veugelers K, Campbell S, Barry M (2009) The fowlpox virus BCL-2 homologue, FPV039, interacts with activated Bax and a discrete subset of BH3-only proteins to inhibit apoptosis. J Virol 83:7085–7098
Taylor JM, Barry M (2006) Near death experiences: poxvirus regulation of apoptotic death. Virology 344:139–150
Taylor JM, Quilty D, Banadyga L, Barry M (2006) The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J Biol Chem 281:39728–39739
Acknowledgments
This work was supported by the Health Research Council of New Zealand and the University of Otago. We wish to thank David Huang and Jamie Fletcher (The Walter and Eliza Hall Institute of Medical Research (WEHI), Australia) for providing information and a range of plasmids expressing Bcl-2 family members. We are grateful to Jerry Adams (WEHI, Australia) for critical reading of the manuscript, Catherine Day and Fabienne Lecomte (University of Otago, New Zealand) for helpful discussions and Ellena Whelan (University of Otago) for technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Elizabeth C. Ledgerwood and Andrew A. Mercer contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Westphal, D., Ledgerwood, E.C., Tyndall, J.D.A. et al. The orf virus inhibitor of apoptosis functions in a Bcl-2-like manner, binding and neutralizing a set of BH3-only proteins and active Bax. Apoptosis 14, 1317–1330 (2009). https://doi.org/10.1007/s10495-009-0403-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-009-0403-1