
Abstract
Apoptosis, the most common form of cell death, is a key mechanism in the build up and maintenance of both innate and adaptive immunity. Central to the apoptotic process is a family of intracellular cysteine proteases with aspartate-specificity, called caspases. Caspases are counter-regulated by multiple anti-apoptotic molecules, and the expression of the latter in leukocytes is largely dependent on survival factors. Therefore, the physiologic rates of apoptosis change under pathologic conditions. For instance, in inflammation, the expression of survival factors is usually elevated, resulting in increased cell survival and consequently in the accumulation of the involved immune cells. In many allergic diseases, eosinophil apoptosis is delayed contributing to both blood and tissue eosinophilia. Besides eosinophils, apoptosis of other leukocytes is also frequently prevented or delayed during allergic inflammatory processes. In contrast to inflammatory cells, accelerated cell death is often observed in epithelial cells, a mechanism, which amplifies or at least maintains allergic inflammation. In conclusion, deregulated cell death is a common phenomenon of allergic diseases that likely plays an important role in their pathogenesis. Whether the apoptosis is too little or too much depends on the cell type. In this review, we discuss the regulation of the lifespan of the participating leukocytes in allergic inflammatory responses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Allergic diseases are the consequence of hyperreactivity reactions of the immune systems towards exogeneous antigens (allergens). Most allergic reactions are associated with an increased T helper (Th) 2 activation, resulting in elevated IgE levels and eosinophilia. Allergic diseases, including asthma, rhinitis, conjunctivitiy, atopic eczema, as well as food and drug allergies are major contributors to morbidity in the civilized world, and sometimes even cause mortality. With the expanding knowledge, the field of allergy, along with the broader framework of immunology, has dramatically changed concepts in recent years. However, in spite of the progress, the regulation of cell death in allergic diseases is relatively little investigated and its contribution to the pathogenesis of the different disorders not well understood and reflected, respectively.
Similar to other types of inflammatory responses, accumulation of subgroups of leukocytes occur during the initiation and maintenance phases, whereas inflammatory cell numbers decline in the resolution phase of allergic inflammation. The changes in cell numbers during inflammation are largely due to changes of rates of both cell generation and cell death. Important leukocyte subgroups believed to play critical roles in the pathophysiology of allergic inflammation involve dendritic cells, T cells, mast cells, and eosinophils. The most common form of cell death of leukocytes is apoptosis.
That apoptosis is deregulated in allergic diseases is often not reflected in reviews dealing with the pathophysiologic relevance of apoptosis in diseases. However, since approximately one-third of the population within the countries of the industrial world suffers from one or several allergic diseases [1], it is important that apoptosis research in these disorders is not neglected. Here, we review the current knowledge on the regulation of cell death during allergic inflammatory responses. Due to space limitations, we will concentrate on summarizing important molecular events that change apoptosis rates in allergic diseases and will not cover basic knowledge on apoptosis pathways.
Cell death of mast cells
Mast cells are resident in all normal tissues, where they are believed to play an important role in tissue homeostasis, wound healing, and host defense. Mast cell activation is a characteristic feature of allergic responses, leading to the release of a large number of important mediators that can cause chronic inflammation. Mast cells are also involved in the mechanisms leading to bronchial hyperreactivity [2]. Therefore, how mast cell homeostasis is regulated may have significant effects on normal physiology and contributes to the genesis of allergic inflammatory diseases.
Mast cell numbers are increased in allergic inflammatory responses [3], and preventing apoptosis might contribute to this phenomenon. Stem cell factor (SCF), interleukin (IL)-3, IL-4, IL-5, IL-6, and nerve growth factor (NGF) have been described to promote mast cell survival [4–6]. On the other hand, it has been reported that exposure of murine bone-marrow-derived mast cells to IL-3, IL-4, and IL-10 downregulate the SCF receptor kit as well as the high-affinity IgE receptor, resulting in mast cell apoptosis [7].
SCF is considered as being the most crucial survival factor for mast cells. The crucial role of SCF for regulating mast cell numbers is best reflected by experimental in vivo models. Mice with deficient expression of SCF or kit have almost complete lack of mast cells in their tissues [8, 9]. SCF is secreted from several cell types, such as stromal cells, fibroblasts, endothelial cells, and mast cells themselves [10, 11]. Injection of SCF to the skin increases mast cell numbers. Moreover, a gain of functional mutations of kit causes systemic mastocytosis [12].
Following binding of SCF to kit, kit is dimerized and autophosphorylated on tyrosine residues. This initiates multiple intracellular signaling pathways, which involve phosphatidylinositol-3-kinase (PI3K), mitogen activated protein kinase (MAPK), phospholipase C γ (PLCγ), Src kinase, and Janus kinase/Signal transducers and activators of transcription (Jak/STAT), resulting in gene activation [13]. It was found that Bim is both inactivated and reduced due to SCF stimulation of mast cells [14]. Interestingly, cross-linking of the high-affinity IgE receptor also leads to apoptosis inhibition, which is mediated by increased Bcl-xL and A1 expression [15, 16]. Increased survival following mast cell activation may also be the consequence of the release of survival factors, which then act in an autocrine manner [17, 18].
Besides the anti-apoptotic mechanisms, mast cells additionally carry functional death receptors. For instance, mast cell apoptosis can be induced following ligation of Fas and TNF-related apoptosis inducing ligand (TRAIL) receptors [19, 20]. Interestingly, immunoglobulin E (IgE)-mediated activation of mast cells increased their sensitivity to undergo TRAIL-induced apoptosis, although the mechanism(s) responsible for these functional effects remain to be investigated [20, 21]. In the resolution phase of allergic inflammation, in which survival cytokine expression is likely decreased, the induction of mast cell apoptosis critically involves the BH3-only protein Puma [22].
Cell death of eosinophils, neutrophils, and basophils
Eosinophils are prominent effector cells in many allergic and parasitic inflammatory responses [23]. They are constantly generated in the bone-marrow and short-lived [24]. Moreover, eosinophils are relatively rare and their contribution to blood leukocyte numbers does not exceed 4% under physiologic conditions. IL-5 represents a crucial cytokine for eosinophil differentiation, activation, and survival [25]. Therefore, in diseases with elevated levels of IL-5, increased numbers of eosinophils are observed [26]. The importance of IL-5 for delayed eosinophil apoptosis in tissues has been directly demonstrated in nasal polyp explants [27]. Delayed eosinophil apoptosis has also been demontrated in experimental in vivo models of allergic disease [28]. Moreover, there was an inverse correlation between numbers of apoptotic eosinophils in sputum from asthmatic patients and levels of IL-5 and eotaxin, again indicating that IL-5 (and perhaps eotaxin) acts as an eosinophil survival factor in vivo [29]. Besides IL-5, other eosinophil survival cytokines are IL-3 and granulocyte/macrophage colony-stimulating factor (GM-CSF). Interestingly, eotaxin-1 [30], leptin [31], and CD40 ligand [32] are also able to prolong eosinophil survival in vitro.
The molecular mechanisms involved in cytokine-mediated enhanced eosinophil survival include increased expression of Bcl-xL [33], delayed Bid cleavage [34], inhibition of Bax translocation to mitochondria [35], and delayed Bax cleavage [31], resulting in delayed mitochondrial cytochrome c and second mitochondria-derived activator of caspase (Smac) release and caspase activation [31, 35]. IL-5 has also been shown to induce cIAP2 and survivin, suggesting that delay of apoptosis can also be achieved by blocking caspases [36]. The signal transduction mechanisms leading to gene expression of anti-apoptotic proteins involve tyrosine kinases [37, 38], MAPK [37], PI3K [39], Jak/STAT [40, 41], and NF-κB pathways [42]. In contrast, the c-Jun N-terminal kinase (JNK) pathway seems to mediate constitutive eosinophil apoptosis [43].
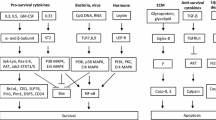
Eosinophils also carry functional death receptors. For instance, eosinophils express Fas receptors that initiate apoptosis upon activation, even in the presence of survival cytokines [44]. In contrast, nitric oxide, that is increasingly generated during allergic inflammatory responses, counterregulates Fas receptor-induced apoptosis [45]. TRAIL and TNF receptors are also expressed on eosinophils, and were reported to mediate eosinophil survival [46, 47]. In contrast, recombinant surfactant protein-D (SP-D) has been shown to induce apoptosis in eosinophils from allergic patients, but not from healthy donors, although the mechanism remains unclear [48]. Eosinophil apoptosis can also be induced as a consequence of sialic acid binding immunoglobulin-like lectin (Siglec)-8 ligation [49]. Moreover, IL-5 increases Siglec-8 mediated death in a partially caspase-independent manner (Fig. 1) [50]. Therefore, the eosinophil cell death under these condition is unlikely apoptosis, but may represent an autophagic-like cell death [51]. Recently, a natural ligand for Siglec-8 has been identified [52]. Interestingly, Siglec-8 can also be ligated by physiologic anti-Siglec-8 autoantibodies [53]. Moreover, ligation of mouse Siglec-F, the closest functional paralog of human Siglec-8, selectively reduces blood and tissue eosinophils in experimental mouse models [54].

Different forms of cell death in eosinophils following Fas and Siglec-8 ligation in the presence of IL-5. Both Fas and Siglec-8 ligation induce caspase activation and ROS generation in the absence of concurrent survival cytokine stimulation, leading to apoptosis. In the presence of IL-5, however, death programs are different. In contrast to Fas ligation, which blocks anti-apoptotic signaling, Siglec-8 ligation does not prevent IL-5-mediated caspase inactivation. Caspase inhibition is associated with a largely caspase-independent cell death, which depends on ROS
Glucocorticoids that are often used as anti-inflammatory drugs in allergic inflammatory responses directly induce eosinophil apoptosis, although the molecular mechanism of this drug’s action on cell survival remains unclear [55]. Nevertheless, this effect might be important since asthmatic patients exhibit an increased proportion of apoptotic eosinophils in their airway secretions following clinical improvement with successful glucocorticoid therapy [56]. Theophylline has also been reported to induce eosinophil apoptosis [57], but the clinical significance of this finding is unclear. Agents that increase intracellular cAMP may also modify eosinophil apoptosis, depending on the inflammatory cytokine environment [58].
Although neutrophils can be present in bronchial asthma [59], they are usually not dominant in chronic allergic inflammatory responses. However, it is interesting to compare the regulation of apoptosis between eosinophils and neutrophils, since both cell types are granulocytes. Neutrophils and eosinophils express surface molecules, which initiate either survival or death signals. Both cell types respond with enhanced survival when stimulated with GM-CSF, and with enhanced apoptosis following ligation of Fas receptors. Like in eosinophils, Fas stimulation results in neutrophil apoptosis even in the presence of survival factors [60]. On the other hand, there are also surface receptors, which are expressed on either eosinophils or neutrophils. For instance, IL-5 is a specific survival factor for eosinophils, whereas G-CSF specifically prolongs the lifespan of neutrophils. In addition, the complement factor C5a enhances neutrophil [61], but not eosinophil survival (unpublished observation). Siglec-8 transduces death signals in eosinophils, but not neutrophils. In contrast, Siglec-9 is a death receptor on neutrophils that is not expressed on eosinophils [62]. Moreover, the nicotinic acid receptor GPR109A was recently identified on the surface of neutrophils, but not eosinophils [63]. There seem to be also differences regarding the mechanism of cell death regulation in association with DNA release between neutrophils and eosinophils [64, 65]. Interestingly, hypoxia, which induces apoptosis in most cell types, delays apoptosis in neutrophils [66]. The effect of hypoxia on eosinophil apoptosis is unknown.
Apoptosis in eosinophils and neutrophils is also regulated by drugs and/or compounds. Glucocorticoids, which induce eosinophil apoptosis (see above), delay the neutrophil apoptotic program [55]. On the other hand, nitric oxide donors promote neutrophil apoptosis, but somehow block eosinophil apoptosis [45]. Furthermore, the effect of phenylarsine oxide on apoptosis is different in neutrophils and eosinophils at a given concentration [67]. These data suggest that differences exist in the expression of intracellular components of cell death pathways between eosinophils and neutrophils. Indeed, caspases, although present, are somehow more difficult to activate in eosinophils compared with neutrophils [68]. In contrast, the expression of Bcl-2 family members seems to be similar in eosinophils and neutrophils. Interestingly, Bcl-2, although present in immature precursors, is no longer present in both granulocyte types upon full maturation [69]. This observation may, at least partially, explain the short lifespan of these cells. Bim, a BH3-only protein, seems to play a major role in the regulation of neutrophil apoptosis [70]. The expression and function of Bim in eosinophils remains to be investigated. Taken together, the regulation of apoptosis in eosinophils and neutrophils is at least partially different, providing the opportunity to selectively target one granulocyte type without affecting the other by pharmacological means.
In contrast to eosinophils and neutrophils, little is known about the regulation of apoptosis in basophils, which represent the least abundant granulocyte population. Basophils are considered, besides mast cells and eosinophils, as key effector cells in allergic inflammation. Proinflammatory and immunomodulatory activities of basophils include secretion of histamine and the lipid mediator leukotriene C4 as well as rapid production of IL-4 and IL-13, Th2-type cytokines crucial for the development of allergy. The regulation of basophil apoptosis is likely to be important for the length and strength of allergic inflammation, since negative regulators on basophil activity have not been identified. IL-3 appears to be the only ligand that protects basophil apoptosis with high efficacy [71]. In contrast, ligation of Fas results in basophil apoptosis [72]. A recent study demonstrated that IL-3 mediates its anti-apoptotic effect on basophils through a Pim1-dependent signaling pathway [73].
Cell death of T cells
T cells, in particular T cells producing Th2 cytokines, play an important role in allergic inflammatory responses. For instance, IL-4 and IL-13 enhance IgE production, IL-4, IL-9, and IL-10 enhance mast cell growth; and IL-5 promotes eosinophil accumulation [74]. This Th2 response is the result of clonal expansion of allergen-specific T cells and involves both increased proliferation and inhibition of apoptosis. Although some observations in allergic inflammatory responses suggest prolonged survival of Th2 cells [75], most of our information on the role of T cell apoptosis in immune responses comes from experimental models. Bcl-2 expression is required for the survival of mature, resting T cells [76]. IL-4, IL-6, or IL-7 are required to maintain Bcl-2 and Bcl-xL levels in these cells [77, 78]. Interaction of the T cell receptor (TCR) with major histocompatibility complex (MHC) class II molecules is required to keep memory T cells alive [79].
The antigen-mediated stimulation of T cells results in a change in requirements for survival. Activated T cells produce IL-2 and are dependent on IL-2, and related cytokines, for their survival [77, 78, 80]. IL-2 and related cytokines maintain Bcl-2 and Bcl-xL levels [77, 78], and IL-2 withdrawal requires activation of Bim to induce death [81]. Repeated TCR activation sensitizes T cells to apoptosis, a process known as activation-induced cell death. Upon activation, Th1 cells are initially resistant to Fas ligand-induced death, but they gain sensitivity after several days [82]. This increased susceptibility towards Fas receptor-mediated apoptosis has been attributed to lower levels of FLICE-like inhibitory protein (FLIP) and is IL-2-dependent [83]. In Th2 cells, however, FLIP levels may not decrease, resulting in Fas resistance [84]. Therefore, Th2 predominance in allergic diseases may largely be due to increased Th1 cell apoptosis [85, 86].
In the resolution phase of inflammatory responses, most activated T cells are killed. T cell apoptosis may occur by two mechanisms: (1) by repeated stimulation of the TCR with antigen in conjunction with MHC class II molecules, and (2) by reduction in cytokine levels due to decreased inflammation. The first mechanism requires Fas in Th1 cells, and Fas-deficient patients develop a lymphoproliferative disease [87, 88]. In Th2 cells, which are Fas resistant [84], granzyme B is critical for activation-induced cell death (Fig. 2) [89]. This second mechanism requires Bim [81] and can be blocked by high levels of Bcl-2 [90]. Th2 cell apoptosis in allergic diseases may also be achieved by drug treatment. For instance, calcineurin inhibitors have been described to induce T cell apoptosis in atopic eczema [91].

Distinct T cell receptor (TCR)-mediated death pathways in Th1 and Th2 cells. Whereas Th1 cells gain Fas sensitivity due to IL-2-dependent downregulation of FLIP during an immune response, Th2 cells stay Fas resistant following TCR activation. In Th2 cells, granzyme B is released from lysosomes mediating a caspase-independent cell death
Almost nothing is known about the regulation of apoptosis of regulatory T cells. It is tempting to speculate, however, that dysregulated apoptosis in these cells contributes to the pathogenesis of allergic inflammatory responses. Interestingly, it has been suggested that T regulatory cells induce effector CD4+ T cell apoptosis in an experimental model of inflammatory bowel disease [92]. Therefore, it is possible that regulatory T cells also fulfill their function, at least partially, by mediating effector T cell apoptosis in allergic diseases. Clearly, such a potential mechanism deserves further investigation.
Cell death of dendritic cells
The role of dendritic cells in driving Th2 allergic responses has received considerable attention in recent years. Dendritic cells have direct contact to incoming antigens. In the presence of a danger signal, dendritic cells mature to professional antigen-presenting cells, and interact with naive T cells in draining lymph nodes. Dendritic cells can influence polarization of T cells by the release of cytokines [93] and their expression of costimulatory molecules [94] that are both influenced by the local environment [95]. Dendritic cells have been shown to be essential in the pathogenesis of allergic diseases [96, 97].
The lifespan of mature dendritic cells is thought to be approximately three days [98]. This short time may limit the availability of antigen for T cells, and apoptosis induction in dendritic cells may serve to regulate immune responses. The lifespan of dendritic cells is determined by both antigen-mediated and T cell signals. For instance, ligands for Toll-like receptors (TLRs), CD40 ligand, or tumor necrosis factor-related activation-induced cytokine (TRANCE) promote dendritic cell survival via NF-κB pathways [99–101]. One of the NF-κB target genes is Bcl-xL, which is induced by both TLR ligands and T cell signals. In addition, TLR ligands, but not T cell signals, reduce Bcl-2 and induce Bim, thus limiting the lifespan of dendritic cells [102]. One might, therefore, speculate that immature dendritic cells receive first TLR ligands, which are likely to set the lifespan of dendritic cells, before they even enter the lymph node. A function of T cells might then be to prolong the survival of dendritic cells, possibly leading to a temporary and local enrichment of dendritic cells.
It is has been demonstrated that increasing the lifespan of mature dendritic cells is an important factor to strengthen the inflammatory response under in vivo conditions [102]. Therefore, it is likely that increased dendritic cell survival also plays a role in allergic inflammatory responses. Indeed, it has recently been reported that apoptosis-resistant dendritic cells promote Th2 responses, including IgE production in an experimental mouse model [103]. Therapeutic approaches promoting dendritic cell apoptosis appear to be promising [104], further pointing to the possibility that the lifespan of dendritic cells is a critical element in the generation and/or maintenance of allergic diseases.
Cell death of epithelial cells
Epithelial cell apoptosis is a common phenomenon of allergic inflammation. For instance, bronchial epithelial apoptosis leads to epithelial shedding in asthma. Apoptosis is mediated by activated T cell and eosinophils [105]. Another example of epithelial cell apoptosis in association with allergy is seen in atopic eczema. Apoptosis of keratinocytes has been reported to be a major cause of spongiosis, which represents one of the hallmarks of atopic eczema [106]. IFN-γ, even at very low concentrations, increases Fas expression on keratinocytes and renders these cells susceptible to apoptosis [106]. Taken together, allergic diseases are often associated with epithelial cell damage, which likely amplifies or at least maintains the inflammatory process.
Conclusion
A look at the molecular basis of many allergic diseases reveals a cell death component that either accounts for the disease or contributes to disease progression. Therefore, current and future anti-allergic therapies should also be analyzed in respect to their effects on cell death pathways.
References
Asher MI, Montefort S, Bjorksten B et al (2006) Worldwide time trends in the prevalence and symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC phases one and three repeat multicountry cross-sectional surveys. Lancet 368:733–743. doi:10.1016/S0140-6736(06)69283-0
Bradding P, Walls AF, Holgate ST (2006) The role of mast cells in the pathophysiology of asthma. J Allergy Clin Immunol 117:1277–1284. doi:10.1016/j.jaci.2006.02.039
Gibson PG, Allen CJ, Yang JP et al (1993) Intraepithelial mast cells in allergic and nonallergic asthma: assessment using bronchial brushings. Am Rev Respir Dis 148:80–86
Mekori YA, Oh CK, Metcalfe DD (1993) IL-3-dependent murine mast cells undergo apoptosis on removal of IL-3: prevention of apoptosis by c-kit ligand. J Immunol 151:3775–3784
Iemura A, Tsai M, Ando A, Wershil BK, Galli SJ (1994) The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am J Pathol 144:321–328
Yanagida M, Fukamachi H, Ohgami K et al (1995) Effects of T-helper 2-type cytokines, interleukin-3 (IL-3), IL-4, IL-5, and IL-6 on the survival of cultured human mast cells. Blood 86:3705–3714
Shelburne CP, Ryan JJ (2001) The role of Th2 cytokines in mast cell homeostasis. Immunol Rev 179:82–93. doi:10.1034/j.1600-065X.2001.790109.x
Kitamura Y, Go S, Hatanaka K (1978) Decrease of mast cells in W/Wv mice and their increase by bone-marrow transplantation. Blood 52:447–452
Kitumara Y, Go S (1979) Decreased production of mast cells in S1/S1d anemic mice. Blood 53:492–497
Heinrich MC, Dooley DC, Freed AC et al (1993) Constitutive expression of steel factor gene by human stromal cells. Blood 82:771–783
Zhang S, Anderson DF, Bradding P et al (1998) Human mast cells express stem cell factor. J Pathol 186:59–66. doi:10.1002/(SICI)1096-9896(199809)186:1<59::AID-PATH140>3.0.CO;2-J
Wershil BK, Tsai M, Geissler EN, Zsebo KM, Galli SJ (1992) The rat c-kit ligand, stem cell factor, induces c-kit receptor-dependent mouse mast cell activation in vivo: evidence that signaling through the c-kit receptor can induce expression of cellular function. J Exp Med 175:245–255. doi:10.1084/jem.175.1.245
Reber L, Da Silva CA, Frossard N (2006) Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur J Pharmacol 533:327–340. doi:10.1016/j.ejphar.2005.12.067
Möller C, Alfredsson J, Engström M et al (2005) Stem cell factor promotes mast cell survival via inactivation of FOXO3a-mediated transcriptional induction and MEK-regulated phosphorylation of the proapoptotic protein Bim. Blood 106:1330–1336. doi:10.1182/blood-2004-12-4792
Asai K, Kitaura J, Kawakami Y et al (2001) Regulation of mast cell survival by IgE. Immunity 14:791–800. doi:10.1016/S1074-7613(01)00157-1
Alfredsson J, Puthalakath H, Martin H, Strasser A, Nilsson G (2005) Proapoptotic Bcl-2 family member Bim is involved in the control of mast cell survival and is induced together with Bcl-xL upon IgE-receptor activation. Cell Death Differ 12:136–144. doi:10.1038/sj.cdd.4401537
Kalesnikoff J, Huber M, Lam V et al (2001) Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity 14:801–811. doi:10.1016/S1074-7613(01)00159-5
Kitaura J, Song J, Tsai M et al (2003) Evidence that IgE molecules mediate a spectrum of effects on mast cell survival and activation via aggregation of the FcepsilonRI. Proc Natl Acad Sci USA 100:12911–12916. doi:10.1073/pnas.1735525100
Hartmann K, Wagelie-Steffen AL, von Stebut E, Metcalfe DD (1997) Fas (CD95, APO-1) antigen expression and function in murine mast cells. J Immunol 159:4006–4014
Berent-Maoz B, Piliponsky AM, Daigle I, Simon HU, Levi-Schaffer F (2006) Human mast cells undergo TRAIL-induced apoptosis. J Immunol 176:2272–2278
Berent-Maoz B, Salemi S, Mankuta D, Simon HU, Levi-Schaffer F (2008) TRAIL mediated signaling in human mast cells: the influence of IgE-dependent activation. Allergy 63:333–340. doi:10.1111/j.1398-9995.2007.01598.x
Ekoff M, Kaufmann T, Engström M et al (2007) The BH3-only protein Puma plays an essential role in cytokine deprivation induced apoptosis of mast cells. Blood 110:3209–3217. doi:10.1182/blood-2007-02-073957
Simon D, Simon HU (2007) Eosinophilic disorders. J Allergy Clin Immunol 119:1291–1300. doi:10.1016/j.jaci.2007.02.010
Rothenberg ME, Hogan SP (2006) The eosinophil. Annu Rev Immunol 24:147–174. doi:10.1146/annurev.immunol.24.021605.090720
Sanderson CJ (1992) Interleukin-5, eosinophils, and disease. Blood 79:3101–3109
Owen WF, Rothenberg ME, Petersen J et al (1989) Interleukin 5 and phenotypically altered eosinophils in the blood of patients with the idiopathic hypereosinophilic syndrome. J Exp Med 170:343–348. doi:10.1084/jem.170.1.343
Simon HU, Yousefi S, Schranz C, Schapowal A, Bachert C, Blaser K (1997) Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J Immunol 158:3902–3908
Tumes DJ, Cormie J, Calvert MG et al (2007) Strain-dependent resistance to allergen-induced lung pathophysiology in mice correlates with rate of apoptosis of lung-derived eosinophils. J Leukoc Biol 81:1362–1373. doi:10.1189/jlb.0106046
Xu J, Jiang F, Nayeri F, Zetterström O (2007) Apoptotic eosinophils in sputum from asthmatic patients correlate negatively with levels of IL-5 and eotaxin. Respir Med 101:1447–1454. doi:10.1016/j.rmed.2007.01.026
Farahi N, Cowburn AS, Upton PD et al (2007) Eotaxin-1/CC chemokine ligand 11: A novel eosinophil survival factor secreted by human pulmonary artery endothelial cells. J Immunol 179:1264–1273
Conus S, Bruno A, Simon HU (2005) Leptin is an eosinophil survival factor. J Allergy Clin Immunol 116:1228–1234. doi:10.1016/j.jaci.2005.09.003
Bureau F, Seumois G, Jaspar F et al (2002) CD40 engagement enhances eosinophil survival through induction of cellular inhibitor of apoptosis protein 2 expression: possible involvement in allergic inflammation. J Allergy Clin Immunol 110:443–449. doi:10.1067/mai.2002.126781
Dibbert B, Daigle I, Braun D et al (1998) Role for Bcl-xL in delayed eosinophil apoptosis by granulocyte-macrophage colony-stimulating factor and interleukin-5. Blood 92:778–783
Segal M, Niazi S, Simons MP, Galati SA, Zangrilli JG (2007) Bid activation during induction of extrinsic and intrinsic apoptosis in eosinophils. Immunol Cell Biol 85:518–524. doi:10.1038/sj.icb.7100075
Dewson G, Cohen GM, Wardlaw AJ (2001) Interleukin-5 inhibits translocation of Bax to mitochondria, cytochrome c release, and activation of caspases in human eosinophils. Blood 98:2239–2247. doi:10.1182/blood.V98.7.2239
Vassina EM, Yousefi S, Simon D, Zwicky C, Conus S, Simon HU (2006) cIAP-2 and survivin contribute to cytokine-mediated delayed eosinophil apoptosis. Eur J Immunol 36:1975–1984. doi:10.1002/eji.200635943
Pazdrak K, Schreiber D, Forsythe P, Justement L, Alam R (1995) The signal transduction mechanism of IL-5 in eosinophils: the involvement of lyn tyrosine kinase and the ras-raf 1-MEK-MAP kinase pathway. J Exp Med 181:1827–1834. doi:10.1084/jem.181.5.1827
Yousefi S, Hoessli DC, Blaser K, Mills GB, Simon HU (1996) Requirement of Lyn and Syk tyrosine kinases for the prevention of apoptosis by cytokines in human eosinophils. J Exp Med 183:1407–1414. doi:10.1084/jem.183.4.1407
Pinho V, Souza DG, Barsante MM et al (2005) Phosphoinositide-3 kinases critically regulate the recruitment and survival of eosinophils in vivo: importance for the resolution of allergic inflammation. J Leukoc Biol 77:800–810. doi:10.1189/jlb.0704386
van der Bruggen T, Caldenhoven E, Kanters D et al (1995) Interleukin-5 signaling in human eosinophils involves JAK2 tyrosine kinase and STAT1α. Blood 85:1442–1448
Simon HU, Yousefi S, Dibbert B, Levi-Schaffer F, Blaser K (1997) Anti-apoptotic signals of granulocyte-macrophage colony-stimulating factor are transduced via Jak2 tyrosine kinase in eosinophils. Eur J Immunol 27:3536–3539. doi:10.1002/eji.1830271256
Fujihara S, Jaffray E, Farrow SN, Rossi AG, Haslett C, Hay RT (2005) Inhibition of NF-kappa B by a cell permeable form of I kappa B alpha induces apoptosis in eosinophils. Biochem Biophys Res Commun 326:632–637. doi:10.1016/j.bbrc.2004.11.090
Hasala H, Zhang X, Saarelainen S, Moilanen E, Kankaanranta H (2007) c-Jun N-terminal kinase mediates constitutive human eosinophil apoptosis. Pulm Pharmacol Ther 20:580–587. doi:10.1016/j.pupt.2006.06.004
Matsumoto K, Schleimer RP, Saito H, Iikura Y, Bochner BS (1995) Induction of apoptosis in human eosinophils by anti-Fas antibody treatment in vitro. Blood 86:1437–1443
Hebestreit H, Dibbert B, Balatti I et al (1998) Disruption of Fas receptor signaling by nitric oxide in eosinophils. J Exp Med 187:415–425. doi:10.1084/jem.187.3.415
Daigle I, Simon HU (2001) Alternative functions for TRAIL receptors in eosinophils and neutrophils. Swiss Med Wkly 131:231–237
Temkin V, Levi-Schaffer F (2001) Mechanisms of tumour necrosis factor alpha mediated eosinophil survival. Cytokine 15:20–26. doi:10.1006/cyto.2001.0890
Mahajan L, Madan T, Kamal N et al (2008) Recombinant surfactant protein-D selectively increases apoptosis in eosinophils of allergic asthmatics and enhances uptake of apoptotic eosinophils by macrophages. Int Immunol 20:993–1007. doi:10.1093/intimm/dxn058
Nutku E, Aizawa H, Hudson SA, Bochner BS (2003) Ligation of Siglec-8: a selective mechanism for induction of human eosinophil apoptosis. Blood 101:5014–5020. doi:10.1182/blood-2002-10-3058
Nutku-Bilir E, Hudson SA, Bochner BS (2008) Interleukin-5 priming of human eosinophils alters Siglec-8 mediated apoptosis pathways. Am J Respir Cell Mol Biol 38:121–124. doi:10.1165/rcmb.2007-0154OC
von Gunten S, Simon HU (2007) Autophagic-like cell death in neutrophils induced by autoantibodies. Autophagy 3:67–68
Bochner BS, Alvarez RA, Mehta P et al (2005) Glycan array screening reveals a candidate ligand for Siglec-8. J Biol Chem 280:4307–4312. doi:10.1074/jbc.M412378200
von Gunten S, Vogel M, Schaub A et al (2007) Intravenous immunoglobulin preparations contain anti-Siglec-8 autoantibodies. J Allergy Clin Immunol 119:1005–1011. doi:10.1016/j.jaci.2007.01.023
Zimmermann N, McBride ML, Yamada Y et al (2008) Siglec-F antibody administration to mice selectively reduces blood and tissue eosinophils. Allergy 63:1156–1163. doi:10.1111/j.1398-9995.2008.01709.x
Meagher LC, Cousin JM, Seckl JR, Haslett C (1996) Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J Immunol 156:4422–4428
Woolley KL, Gibson PG, Carty K, Wilson AJ, Twaddell SH, Woolley MJ (1996) Eosinophil apoptosis and the resolution of airway inflammation in asthma. Am J Respir Crit Care Med 154:237–243
Yasui K, Hu B, Nakazawa T, Agematsu K, Komiyama A (1997) Theophylline accelerates human granulocyte apoptosis not via phosphodiesterase inhibition. J Clin Invest 100:1677–1684. doi:10.1172/JCI119692
Hallsworth MP, Giembycz MA, Barnes PJ, Lee TH (1996) Cyclic AMP-elevating agents prolong or inhibit eosinophil survival depending on prior exposure to GM-CSF. Br J Pharmacol 117:79–86
Shaw DE, Berry MA, Hargadon B et al (2007) Association between neutrophilic airway inflammation and airflow limitation in adults with asthma. Chest 132:1871–1875. doi:10.1378/chest.07-1047
Daigle I, Yousefi S, Colonna M, Green DR, Simon HU (2002) Death receptors bind SHP-1 and block cytokine-induced anti-apoptotic signalling in neutrophils. Nat Med 8:61–67. doi:10.1038/nm0102-61
Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL (2002) C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int 61:456–463. doi:10.1046/j.1523-1755.2002.00139.x
von Gunten S, Yousefi S, Seitz M et al (2005) Siglec-9 transduces apoptotic and nonapoptotic death signals into neutrophils depending on the proinflammatory cytokine environment. Blood 106:1423–1431. doi:10.1182/blood-2004-10-4112
Kostylina G, Simon D, Fey MF, Yousefi S, Simon HU (2008) Neutrophil apoptosis mediated by nicotinic acid receptors (GPR109A). Cell Death Differ 15:134–142. doi:10.1038/sj.cdd.4402238
Fuchs TA, Abed U, Goosmann C et al (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–241. doi:10.1083/jcb.200606027
Yousefi S, Gold JA, Andina N et al (2008) Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14:949–953. doi:10.1038/nm.1855
Walmsley SR, Print C, Farahi N et al (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J Exp Med 201:105–115. doi:10.1084/jem.20040624
Yousefi S, Green DR, Blaser K, Simon HU (1994) Protein-tyrosine phosphorylation regulates apoptosis in human eosinophils and neutrophils. Proc Natl Acad Sci USA 91:10868–10872. doi:10.1073/pnas.91.23.10868
Daigle I, Simon HU (2001) Critical role for caspases 3 and 8 in neutrophil but not eosinophil apoptosis. Int Arch Allergy Immunol 126:147–156. doi:10.1159/000049506
Altznauer F, Martinelli S, Yousefi S et al (2004) Inflammation-associated cell cycle-independent block of apoptosis by survivin in terminally differentiated neutrophils. J Exp Med 199:1343–1354. doi:10.1084/jem.20032033
Villunger A, Scott C, Bouillet P, Strasser A (2003) Essential role for the BH3-only protein Bim, but redundant roles for Bax, Bcl-2 and Bcl-w in the control of granulocyte survival. Blood 101:2393–2400. doi:10.1182/blood-2002-07-2132
Zheng X, Karsan A, Duronio V et al (2002) Interleukin-3, but not granulocyte-macrophage colony-stimulating factor and interleukin-5 inhibits apoptosis of human basophils through phosphatidylinositol 3-kinase: requirement of NF-κB-dependent and -independent pathways. Immunol 107:306–315. doi:10.1046/j.1365-2567.2002.01517.x
Matsumoto K, Maeda A, Bochner BS, Wakiguchi H, Saito H (2008) Induction of apoptosis in human basophils by anti-Fas antibody treatment in vitro. Int Arch Allergy Immunol 146(suppl. 1):40–46. doi:10.1159/000126060
Didichenko SA, Spiegl N, Brunner T, Dahinden CA (2008) IL-3 induces a Pim1-dependent anti-apoptotic pathway in primary human basophils. Blood. doi:10.1182/blood-2008-04-149419
Umetsu DT, DeKruyff RH (2006) The regulation of allergy and asthma. Immunol Rev 212:238–255. doi:10.1111/j.0105-2896.2006.00413.x
Akdis CA, Blaser K, Akdis M (2004) Apoptosis in tissue inflammation and allergic disease. Curr Opin Immunol 16:717–723. doi:10.1016/j.coi.2004.09.004
Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ (1993) Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75:229–240. doi:10.1016/0092-8674(93)80065-M
Vella AT, Dow S, Potter TA, Kappler J, Marrack P (1998) Cytokine-induced survival of activated T cells in vitro and in vivo. Proc Natl Acad Sci USA 95:3810–3815. doi:10.1073/pnas.95.7.3810
Li XC, Demirci G, Ferrari-Lacraz S et al (2001) IL-15 and IL-2: a matter of life and death for T cells in vivo. Nat Med 7:114–118. doi:10.1038/83253
Kirberg J, Berns A, von Boehmer H (1997) Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex-encoded molecules. J Exp Med 186:1269–1275. doi:10.1084/jem.186.8.1269
Duke RC, Cohen JJ (1986) IL-2 addiction: withdrawal of growth factor activates a suicide program in dependent T cells. Lymphokine Res 5:289–299
Hildeman DA, Zhu Y, Mitchell TC et al (2002) Activated T cell death in vivo mediated by pro-apoptotic Bcl-2 family member Bim. Immunity 16:759–767. doi:10.1016/S1074-7613(02)00322-9
Arnold R, Brenner D, Becker M, Frey CR, Krammer PH (2006) How T lymphocytes switch between life and death. Eur J Immunol 36:1654–1658. doi:10.1002/eji.200636197
Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK (1998) Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 8:615–623. doi:10.1016/S1074-7613(00)80566-X
Zhang XR, Zhang LY, Devadas S, Li L, Keegan AD, Shi YF (2003) Reciprocal expression of TRAIL and CD95L in Th1 and Th2 cells: role of apoptosis in T helper subset differentiation. Cell Death Differ 10:203–210. doi:10.1038/sj.cdd.4401138
Akdis M, Trautmann A, Klunker S et al (2003) T helper (Th) 2 predominance in atopic disease is due to preferential apoptosis of circulating memory/effector Th1 cells. FASEB J 17:1026–1035. doi:10.1096/fj.02-1070com
Akkoc T, de Koning PJ, Rückert B, Barlan I, Akdis M, Akdis CA (2008) Increased activation-induced cell death of high IFN-γ-producing Th1 cells as a mechanism of Th2 predominance in atopic diseases. J Allergy Clin Immunol 121:652–658. doi:10.1016/j.jaci.2007.12.1171
Rieux-Laucat F, Le Deist F, Hivroz C et al (1995) Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 268:1347–1349. doi:10.1126/science.7539157
Fisher GH, Rosenberg FJ, Straus SE et al (1995) Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81:935–946. doi:10.1016/0092-8674(95)90013-6
Devadas S, Das J, Liu C et al (2006) Granzyme B is critical for T cell receptor-induced cell death of type 2 helper T cells. Immunity 25:237–247. doi:10.1016/j.immuni.2006.06.011
Strasser A, Harris AW, Cory S (1991) Bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 67:889–899. doi:10.1016/0092-8674(91)90362-3
Hoetzenecker W, Ecker R, Kopp T, Stuetz A, Stingl G, Elbe-Bürger A (2005) Pimecrolimus leads to an apoptosis-induced depletion of T cells but not Langerhans cells in patients with atopic dermatitis. J Allergy Clin Immunol 115:1276–1283. doi:10.1016/j.jaci.2005.02.011
Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ (2007) CD4+ CD25+ Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol 8:1353–1362. doi:10.1038/ni1536
Maldonado-Lopez R, Maliszewski C, Urbain J, Moser M (2001) Cytokines regulate the capacity of CD8alpha(+) and CD8alpha(−) dendritic cells to prime Th1/Th2 cells in vivo. J Immunol 167:4345–4350
Harris NL, Ronchese F (1999) The role of B7 costimulation in T-cell immunity. Immunol Cell Biol 77:304–311. doi:10.1046/j.1440-1711.1999.00835.x
Vieira PL, de Jong EC, Wierenga EA, Kapsenberg ML, Kalinski P (2000) Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instructions. J Immunol 164:4507–4512
van Rijt LS, Jung S, Kleinjan A et al (2005) In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med 201:981–991. doi:10.1084/jem.20042311
Kleinjan A, Willart M, van Rijt LS et al (2006) An essential role for dendritic cells in human and experimental allergic rhinitis. J Allergy Clin Immunol 118:1117–1125. doi:10.1016/j.jaci.2006.05.030
Ingulli E, Mondino A, Khoruts A, Jenkins MK (1997) In vivo detection of dendritic cell antigen presentation to CD4+ T cells. J Exp Med 185:2133–2141. doi:10.1084/jem.185.12.2133
Park Y, Lee SW, Sung YC (2002) Cutting edge: CpG DNA inhibits dendritic cell apoptosis by up-regulating cellular inhibitor of apoptosis proteins through the phosphatidylinositide-3′-OH kinase pathway. J Immunol 168:5–8
Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P (1998) Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med 188:2175–2180. doi:10.1084/jem.188.11.2175
Wong BR, Josien R, Lee SY et al (1997) TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med 186:2075–2080. doi:10.1084/jem.186.12.2075
Hou WS, van Parijs L (2004) A Bcl-2-dependent molecular timer regulates the lifespan and immunogenicity of dendritic cells. Nat Immunol 5:583–589. doi:10.1038/ni1071
Arques JL, Regoli M, Bertelli E, Nicoletti C (2008) Persistence of apoptosis-resistant T cell-activating dendritic cell promotes T helper type-2 response and IgE antibody production. Mol Immunol 45:2177–2186. doi:10.1016/j.molimm.2007.12.004
Schuller E, Oppel T, Bornhövd E, Wetzel S, Wollenberg A (2004) Tacrolimus ointment causes inflammatory dendritic epidermal cell depletion but not Langerhans cell apoptosis in patients with atopic dermatitis. J Allergy Clin Immunol 114:137–143. doi:10.1016/j.jaci.2004.03.021
Trautmann A, Schmid-Grendelmeier P, Krüger K et al (2002) T cells and eosinophils cooperate in the induction of bronchial epithelial apoptosis in asthma. J Allergy Clin Immunol 109:329–337. doi:10.1067/mai.2002.121460
Trautmann A, Akdis M, Kleeman D et al (2000) T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenic role in eczematous dermatitis. J Clin Invest 106:25–35. doi:10.1172/JCI9199
Acknowledgments
The laboratory of the authors is supported by the Swiss National Science Foundation (grant no. 310000–107526) and the OPO Foundation (Zurich).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Simon, HU. Cell death in allergic diseases. Apoptosis 14, 439–446 (2009). https://doi.org/10.1007/s10495-008-0299-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0299-1