
Abstract
Betulinic acid (BetA) is a plant-derived pentacyclic triterpenoid that exerts potent anti-cancer effects in vitro and in vivo, but is non toxic to untransformed cells. In our previous study we observed that BetA consistently induced cell death in a broad panel of tumor cell lines. Apoptosis induced by BetA involves activation of caspases, PARP cleavage and DNA fragmentation and was suggested to depend on the mitochondrial pathway. However, conflicting results have been reported with respect to the role of the pro- and anti-apoptotic members of the Bcl-2 family, which are often aberrantly regulated in tumors and thereby confer growth and survival advantages. Here we show that BetA-induced apoptosis critically depends on the release of cytochrome c from the mitochondria and formation of the apoptosome. Nevertheless, over-expression of Bcl-2 or Bcl-XL only provides limited protection against BetA-induced apoptosis. More importantly, Bax/Bak deficient cells are as sensitive to BetA as their wild-type counterparts, suggesting that cytochrome c is released in a non-classical fashion. In agreement, pre-incubation with cyclosporin A indicated a crucial role for the mitochondrial permeability transition pore (PT) in the induction of apoptosis. Our observations therefore indicate that BetA affects mitochondria and induces cytochrome c release directly via PT Pore. This is only temporarily prevented by anti-apoptotic members of the Bcl-2 family, but independent of Bax and Bak. These findings help to explain the remarkable broad efficacy of BetA against tumor cells of different origin and its effect in tumor cells that are resistant to other chemotherapeutic agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Betulinic acid (BetA) is a naturally occurring triterpenoid that has been initially described to specifically kill melanoma cells via induction of apoptosis [1] and was later shown to have specificity for neuroectoderm-derived tumors. However, it is becoming clear that its efficacy extends to many other cancer cell lines derived from a variety of different malignancies such as leukemia, prostate, ovarian, breast, lung, and colon cancer [2–6]. Importantly, BetA-induced apoptosis appears to be independent of p53 [6–8], but does show remarkable selectivity for tumor cells over non-transformed cells.
The extrinsic or death receptor pathway is not involved in BetA-induced apoptosis [9]. However, formation of reactive oxygen species (ROS) and a decrease in the mitochondrial membrane potential have been repeatedly associated with BetA treatment [4, 10, 11], which prompted the hypothesis that BetA induces apoptosis via the mitochondrial pathway [12–14]. This pathway is normally regulated by a carefully balanced interplay between pro- and anti-apoptotic members of the Bcl-2 family. Over-expression of pro-survival molecules, such as Bcl-2, Bcl-XL or Mcl-1 or deletion of pro-apoptotic members, such as Bax and Bak, or alternatively deregulation of BH3-only molecules like Bim or Puma, is often observed in tumors and causes resistance of these cells to intrinsic death stimuli [15].
In agreement with a role for the mitochondria in BetA-induced apoptosis is the observation that over-expression of Bcl-2 or Bcl-XL prevents BetA-induced cytochrome c release, caspase activation and PARP cleavage in SHEP neuroblastoma cells [12, 13]. However, separate studies have shown that BetA rather increases the expression of Bcl-2, but can also modulate the expression of other pro- and anti-apoptotic Bcl-2 family members with distinct outcomes. For instance, Mcl-1 was strongly induced in melanoma cells after treatment with BetA [7], while expression of Bcl-XS and Bax were induced in neuroblastoma cells after treatment with BetA [7, 9]. These conflicting observations make it difficult to assess the role of the Bcl-2 family in BetA-induced apoptosis. Nevertheless, the broad anti-tumor effect of BetA renders it unlikely that induction of apoptosis would strictly depend on the classical Bcl-2 regulated mitochondrial pathway, as this is often disrupted in tumor cells [7, 15]. Previously we have shown that in Jurkat T leukemia cells apoptosis is induced upon incubation with BetA. Both classical and non-classical pathways to apoptosis exist [7, 16, 17] but the mechanism used by BetA remains incompletely defined. We therefore set out to delineate the signaling pathway and specifically the role of the Bcl-2 family regulated pathway in BetA-induced apoptosis in further detail. We show that Bcl-2 does not provide effective protection against BetA-induced apoptosis, especially not at later time points. Moreover, for the first time, we demonstrate that Bax/Bak double deficient mouse embryonic fibroblasts (Bax/Bak DKO MEFs) display cytochrome c release, caspase activation and PARP cleavage upon BetA treatment. This indicates that BetA does not induce a classical mitochondrial pathway to apoptosis. The mitochondria are critically involved though, as inhibition of the mitochondrial permeability transition (PT) pore by cyclosporin A (CsA) in combination with Bcl-2 over-expression provided effective protection from BetA induced apoptosis.
Materials and methods
Chemicals
Betulinic acid (≥99% pure, Bioservice Halle) was dissolved at 4 mg/ml in DMSO and aliquots were stored at −80°C. zVAD.fmk (benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone), etoposide, TMRE (Tetramethylrhodamine ethyl ester perchlorate), cyclosporin A and propidium iodide (PI) were purchased from Sigma–Aldrich. Anti-APO-1 was a kind gift from Dr. Peter Krammer.
Antibodies
For western blot analysis anti-caspase-3 (AF-605-NA, R&D and #9662, Cell Signaling) was used as well as an antibody specific for cleaved capsase-3 (Asp175, Cell Signaling) for mouse embryonic fibroblasts, furthermore anti-PARP (#9542, Cell Signaling), anti caspase-7 (#9492, Cell Signaling), anti-Bak (#06-536, Millipore), anti-Bax (N-20, Santa Cruz), anti Bcl-XL (B22630, BD; SC-634, Santa Cruz), and anti-Bcl-2 (N-19, Santa Cruz) were used. Anti-cytochrome c for FACS staining was obtained from BD (clone 6H2.B4).
Cells
Jurkat cells over-expressing Bcl-2, Bcl-XL and wild-type (wt) control cells were obtained from Dr. Jannie Borst (NKI, Amsterdam), MCF-7/FAS and MCF-7/FAS Bcl-2 or Bcl-XL were obtained from Dr. Marja Jäättelä (Danish Cancer Society, Copenhagen) [18]. FADD-deficient, Casp-8- deficient and control Jurkat cells (JA3) were obtained from Dr. John Blenis (Harvard Medical School, Boston), Bax/Bak DKO MEFs and wild-type control MEFs were from Dr. Stanley Korsmeyer. APAF-1 and Caspase-9 knockout and wild-type MEFs were from Dr. Tak Mak (University of Toronto, Canada) and Dr. Richard Flavell (Yale, New Haven, USA), respectively. SW480 were provided by J. van Eendenburg and Dr. A. Gorter (department of Pathology, Leiden University Medical Center, The Netherlands), A549 were from ATCC, and HCT116 lines were from Dr. G. C. Chinnadurai (St Louis University School of Medicine, USA) and Dr. B. Vogelstein (Johns Hopkins, USA). All cells were cultured in IMDM supplemented with 8% FCS, 2 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin.
DNA fragmentation
Apoptotic DNA fragmentation was measured as previously described [6, 7]. Briefly, cells were resuspended in Nicoletti buffer containing 50 μg/ml PI for a minimum of 24 h. Afterwards DNA content was determined in the resulting nuclei using flow cytometric measurement of PI.
Western blot analysis
Cells were lysed in Triton X-100 buffer on ice and protein was quantified using a BCA kit from PIERCE according to manufactures protocol. A volume of 10–15 μg total protein was loaded per lane for SDS–PAGE and blotted onto PVDF transfer membrane (Amersham Biosciences). Blocking of unspecific binding sites was performed overnight at 4°C in 5% low fat milk powder in PBS/0.2% Tween-20 (blocking buffer), blots were then incubated with the primary antibody for 2 h at room temperature in blocking buffer, washed and incubated in blocking buffer with a secondary, HRP labeled antibody. For chemiluminescent visualization, ECL from Amersham Biosciences was used.
Cytochrome c release by FACS staining
Cytochrome c release assay was performed according to the protocol of Waterhouse [7, 19]. Briefly, cells were trypsinized, washed with PBS and incubated with 50 μg/ml digitonin in PBS with 100 mM KCl for 5–10 min. Permeabilization of the cell membrane was assessed using trypan blue exclusion. When the majority of the cells in the aliquot used for trypan blue staining were penetrated by the dye, cells were fixed in 4% paraformaldehyde in PBS for 30 min, washed and incubated in blocking buffer (3% BSA, 0.05% saponin, 0.02% azide in PBS supplemented with normal goat serum, dilution 1:200) for 1 h at room temparature. Anti-cytochrome c incubation was done overnight at 4°C, cells were washed three times and FITC conjugated secondary antibody was applied for 1 h at 4°C. After another washing step cells were analyzed by flow cytometry.
Assessment of mitochondrial membrane potential
Cells were incubated in growth medium containing 25 nM TMRE for 15–20 min at 37°C, resuspended in HEPES buffer, pH 7.4, containing 25 nM TMRE and analyzed by flow cytometry.
Results
BetA-induced apoptosis depends on the apoptosome
To determine whether apoptosis is induced in a classical caspase-dependent fashion, Jurkat cells were treated with BetA in the presence or absence of the pan-caspase inhibitor zVAD.fmk. In line with previous observations, BetA induced caspase-3 and PARP cleavage as well as DNA fragmentation and all these effects were inhibited in the presence of 20 μM zVAD.fmk (Fig. 1a) [6, 12, 13].

BetA induced apoptosis depends on the apoptosome. a Jurkat cells were pretreated with 20 μM zVAD.fmk for 2 h before addition of 10 μg/ml BetA. After 24 h of treatment cells were subjected to western blot analysis (lanes 1: non treated; 2: Betulinic acid; 3: 5 μg/ml etoposide; 4: 1 μg/ml anti-APO-1). DNA fragmentation of BetA treated cells was monitored after 24, 48, and 72 h. Etoposide and anti-APO-1 were included as a control for caspase-3 and PARP cleavage. b Jurkat control (JA3), FADD deficient (FADD ko), and caspase-8 deficient (casp-8 ko) cells were treated with indicated concentrations of BetA or anti-APO1 (1 μg/ml) for 48 h and subjected to nicoletti analysis. c Wild-type (wt), caspase-9 knockout (C9), and Apaf-1 knockout (A) mouse embryonic fibroblasts (MEF) were treated with 10 or 15 μg/ml BetA for 24 h and caspase-3 and PARP cleavage was analyzed by immunoblotting. * Statistical significant difference (t-test) P < 0.05
The death receptor or extrinsic pathway is not involved in BetA-induced apoptosis [2, 9, 11, 20]. In agreement, Jurkat cells deficient in either FADD (FAS Associated Death Domain) or caspase-8, two crucial mediators in the extrinsic pathway, revealed similar sensitivity to BetA, whereas these cells were fully protected against CD95-induced apoptosis (Fig. 1b). These results suggest that BetA is able to induce caspase activation in a death receptor-independent and therefore likely a mitochondria dependent fashion. To validate whether the apoptosome, the signaling complex for capase-9 activation, is crucial for downstream caspase activation we used cells devoid of a functional apoptosome due to deletion of either Apaf-1 or caspase-9. MEFs lacking caspase-9 or Apaf-1 and wild-type MEFs were treated with various concentrations of BetA and apoptosis induction was analyzed after 24 h. Importantly, processing of both caspase-3 and PARP was blocked in caspase-9 deficient cells and in cells lacking Apaf-1 (Fig. 1c), indicating that the apoptosome is a key platform for downstream caspase activity in BetA-treated cells.
Anti-apoptotic Bcl-2 family members partially prevent BetA-induced apoptosis
Previously, the neuroblastoma cell line SHEP was shown to become resistant to BetA when over-expressing Bcl-2 [9, 12, 13]. This would fit the role of cytochrome c in the induction of apoptosis. In agreement with these observations we detected that BetA-induced DNA fragmentation after 24 h was reduced, albeit partially, by Bcl-2 over-expression in Jurkat cells (Fig. 2b, c). Surprisingly, this protective effect was lost at later time points and after 72 h of treatment DNA fragmentation was induced as effectively in Bcl-2 over-expressing cells as compared to control cells (Fig. 2c). In contrast, etoposide-induced DNA fragmentation was prevented by Bcl-2 at all time points analyzed (Fig. 2a). This indicates that the level of Bcl-2 was sufficient to provide protection against a typical mitochondrial-dependent drug, but that BetA can circumvent this protection and BetA induced apoptosis is only delayed by Bcl-2.

Bcl-2 over-expression delays BetA induced apoptosis in Jurkat cells. a Vector control and Bcl-2 over-expressing cells were treated with 5 μg/ml etoposide for 24, 48, and 72 h and DNA fragmentation was measured. b Vector control (ctr) and Bcl-2 over-expressing (Bcl-2) Jurkat cells were analyzed for Bcl-2 expression by immunoblot analysis. c Vector control (■) and Bcl-2 over-expressing (▲) Jurkat cells were treated with indicated concentrations of BetA for 24, 48, and 72 h and DNA fragmentation was assessed. * Statistical significant difference (t-test) P < 0.05
To further dissect the role of the Bcl-2 family in BetA-induced apoptosis and to generalize these observations we used different lines expressing high levels of anti-apoptotic Bcl-2 family members. The breast cancer cell line MCF-7 has been shown to be relatively resistant to BetA-induced apoptosis as measured by PI exclusion [6] and DNA fragmentation [9]. Nevertheless, MCF-7 cells are as sensitive as other tumor cells when clonogenic survival is measured [6]. MCF-7 lacks functional caspase-3 due to a frameshift mutation [21], which could explain its resistance to BetA-induced apoptosis. Indeed, when we measured DNA fragmentation by means of FACS analysis of propidium iodide stained nuclei, we did not observe a sub-G1 peak, which is indicative for DNA fragmentation (data not shown). To determine whether BetA was incapable of activating caspases in MCF-7 we also analyzed PARP cleavage, which is a general substrate for executioner caspases and is not necessarily impaired in cells lacking caspase-3 [22]. Interestingly, PARP was clearly processed 48 h after BetA addition (Fig. 3a). This is likely due to the activation of caspase-7, which was cleaved after BetA treatment (Fig. 3a). Similar to Jurkat cells, MCF-7 cells over-expressing Bcl-2 or Bcl-XL [18] were also clearly sensitive to BetA-induced caspase-7 cleavage and PARP processing, although partial protection was evident (Fig. 3b). Similarly, DNA fragmentation in Ramos cells over-expressing Bcl-2 or Mcl-1 was not prevented upon BetA treatment (data not shown), which further substantiates our conclusion that anti-apoptotic Bcl-2 family members do not provide effective protection against BetA-induced apoptosis, especially not at later time points.

Bcl-2 or Bcl-XL over-expression partially prevents BetA-induced apoptosis in MCF-7 cells. a MCF-7 cells were treated with 10 μg/ml BetA for 48 h and PARP processing and caspase-7 cleavage was assessed. b MCF-7/FAS, MCF-7/FAS Bcl-2, and MCF-7/FAS Bcl-XL were treated with 10, 15 or 20 μg/ml BetA for 48 h and immunoblot staining was performed to assess caspase-7 and PARP cleavage. Immunoblot staining for capsase-7 and PARP was performed on the same blot
Bax/Bak double deficient cells are sensitive to BetA-induced apoptosis
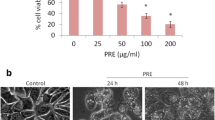
Because expression, interaction and regulation of Bcl-2 family member proteins is complex and can differ in distinct cell types [23, 24] we decided to make use of cells lacking both Bax and Bak. Different models exist describing the interaction between the pro- and anti-apoptotic molecules [25], however, it is agreed upon that Bax or Bak are required for classical cytochrome c release from the mitochondria and subsequent caspase activation [25, 26]. Indeed, MEFs lacking Bax and Bak (Fig. 4a) were resistant to starvation-induced apoptosis and death while wild-type MEFs readily showed caspase-3 activation (Fig. 4b) and morphological cell death (Fig. 4c). To determine if BetA-induced apoptosis was still observed in Bax/Bak DKO, we performed western blot analysis using antibodies specific for PARP and cleaved caspase-3. Strikingly both PARP and caspase-3 were processed as efficiently in the wild-type and Bax/Bak DKO MEFs (Fig. 4d). This suggests that BetA-induced apoptosis is activated in a Bax/Bak-independent, but mitochondria-dependent fashion. To rule out the possibility that these findings are selective for MEFs we also obtained HCT116 colon cancer cells that were made deficient for both Bax and Bak [27, 28]. Similar to the MEFs, we observed that BetA-induced apoptosis was readily induced in cells lacking Bax and Bak (Fig. 4e), indicating that BetA induces apoptosis independent of these pro-apoptotic Bcl-2 family members.

Bax/Bak DKO MEFs are sensitive to BetA-induced apoptosis. a Bax and Bak expression was determined in MEFs by immunoblot analysis. b Cells were functionally tested by washing three times with PBS and incubation in medium without serum (no FCS) for 24 h and subsequently tested for the presence of cleaved caspase-3 (casp-3 cleaved). c Wild-type (wt) and Bax/Bak double deficient (DKO) mouse embryonic fibroblasts were treated with indicated concentrations of BetA for 24 h or subjected to FCS withdrawal and photographed under a phase-contrast microscope at ×100 magnification. d Wild-type (wt) and Bax/Bak double deficient (DKO) cells were treated with 10 or 15 μg/ml BetA for 24 h and immunoblot staining for cleaved caspase-3 and PARP processing was performed. ERK was used as a control for equivalent protein loading. e HCT116 CL (Bax KO) and HCT116 DKO (Bax KO and Bak KD) were incubated with 10 or 15 μg/ml BetA and DNA fragmentation was tested with nicoletti. Expression of Bax and Bak were validated by western (not shown)
Cytochrome c is released upon BetA treatment in Bax/Bak DKO MEFs
Although Bax and Bak appear to be dispensable, the apoptosome is crucial for BetA induced apoptosis. Functional apoptosome formation requires the release of cytochrome c from the mitochondria. We therefore analyzed whether cytochrome c was released upon BetA treatment. In untreated wild-type MEFs cytochrome c was detected at high levels in the mitochondria using intracellular FACS analysis (Fig. 5a). Treatment with BetA or FCS starvation for 24 h resulted in the appearance of a second peak that signifies cells that have released cytochrome c from their mitochondria, indicating that BetA and starvation both induce cytochrome c release. Importantly, cytochrome c release was also detected in casp-9 and APAF-1 KO MEFs (data not shown), which shows that their lack of apoptosis induction is not due to a lack of mitochondrial dysfunction. As Bax/Bak DKO MEFs undergo apoptosis as efficiently as wild-type, our results could point to a Bax/Bak-independent release of cytochrome c. In agreement, a similar percentage of Bax/Bak DKO cells displayed cytochrome c release upon BetA treatment (Fig. 5b). It is important to note that the level of cytochrome c retained in the mitochondria of treated cells appeared slightly higher in Bax/Bak DKO cells (Fig. 5a). This indicates that the number of cells with cytochrome c release is the same, but that the amount of release per cell seems to be less, potentially due to a lack of amplification. BetA thus releases cytochrome c in a Bax/Bak-independent fashion.

BetA induces Bax/Bak independent cytochrome c release. Wild-type (wt) and Bax/Bak double deficient (DKO) mouse embryonic fibroblasts (MEFs) were treated for 24 h with indicated concentrations of BetA or subjected to FCS withdrawal and intracellular staining for cytochrome c release was performed. Representative FACS histograms for cytochrome c release are shown (a), release was quantified using CellQuest software (b)
BetA induced mitochondrial depolarization and apoptosis is dependent on the PT pore
Combined these observations indicate that apoptosis induction by BetA is independent of Bax and Bak, but is, at least partially, affected by Bcl-2 or Bcl-XL over-expression. This suggests that the pathway induced by BetA is not a classical mitochondrial Bcl-2 family-dependent one. Previous findings have indicated that under specific conditions, anti-apoptotic Bcl-2 family members can be cleaved and thereby converted into pro-apoptotic molecules directly facilitating cytochrome c release [29–31]. Although such a pathway could explain our current observations, no evidence of cleavage of either Bcl-2 or Bcl-XL was detected upon BetA treatment (Fig. 6a). Moreover, the levels of Bcl-2 and Bcl-XL compared to a control protein ERK-2 were unaffected upon BetA treatment.

BetA induced mitochondrial depolarization and apoptosis is dependent on the PT pore. a BetA treated Jurkat wild-type (wt), Bcl-2 or Bcl-XL (XL) over-expressing cells were lysed after 24 h of treatment and stained with anti Bcl-2 or anti Bcl-XL antibody. Erk is shown as loading control. b Jurkat wild-type cells were pre-treated with 20 μM zVAD-fmk for 1 h before BetA addition (10 μg/ml). After 24 h mitochondrial depolarization was measured using TMRE. c Jurkat wild-type and Bcl-2 over-expressing cells were pre-incubated with or without 5 μg/ml cyclosporin A (CsA) for 45 min and exposed to 10 μg/ml BetA for 24 h. Mitochondrial depolarization was measured using TMRE. Representative FACS histograms are shown and depolarization was quantified using CellQuest software. d Experiment was performed as described above (c) but instead of treatment with BetA, cells were treated with 5 μg/ml etoposide for 24 h. e Jurkat wild-type and Bcl-2 over-expressing cells were pre-treated with or without 5 μg/ml cyclosporin A (CsA) for 45 min and exposed to 10 μg/ml BetA for 24 h and cytochrome c release was assessed using intracellular FACS staining. f Jurkat cells were treated as described above (e) and after 72 h DNA fragmentation was measured. (Statistics: t-test Fig. 6 b; one-way ANOVA Fig. 6 c–f; symbols: ns not significant; *P < 0.05; **P < 0.01)
The role of the mitochondrial permeability transition (PT) pore is controversial in apoptosis signaling. Some results strongly indicate that it is a secondary effect that is observed only upon downstream caspase activation, while other observations rather propose a causal role for the PT [32]. Opening of the PT pore results in membrane depolarization and is suggested to also lead to cytochrome c release plus subsequent apoptosis. The composition of the pore that plays a role in apoptosis is still a matter of debate. Initial observations indicated that the pore consists of VDAC, ANT, and cyclophilin D [33]. However, recent findings on knockouts lacking all ANT or VDAC forms suggest a more complicated picture [34, 35], at least when looking at the role of this pore in apoptosis. Nonetheless, induction of PT can be directly affected by Bcl-2 family members [36]. We therefore tested the role of PT using TMRE to measure the mitochondrial membrane potential. We found that BetA induced a significant mitochondrial depolarization in Jurkat cells after 24 h, which was independent of caspase activation (Fig. 6b). Importantly, depolarization was reduced in cells over-expressing Bcl-2 (Fig. 6c). Etoposide, a more classical mitochondria-dependent compound, induced depolarization even more dramatic in control cells, but this effect was completely blocked by Bcl-2 (Fig. 6d) and to some extent caspase-dependent (not shown). This again points to the fact that BetA utilizes a different mechanism to target the mitochondria. To more directly analyze the involvement of the PT pore in BetA-induced apoptosis we made use of cyclosporin A (CsA), an inhibitor of cyclophilin D that prevents opening of the pore and thereby depolarization [36]. In agreement, CsA reduced BetA-induced loss of TMRE staining by almost 50% in Jurkat control cells and completely prevented mitochondrial depolarization (Fig. 6c) and cytochrome c release (Fig. 6e) in Jurkat-Bcl-2 cells. In contrast, CsA did not have any protective effects on etoposide treated cells (Fig. 6d). More importantly, apoptosis as measured by DNA fragmentation after 72 h of BetA treatment was substantially inhibited by CsA alone and almost completely when it was combined with Bcl-2 over-expression (Fig. 6f).
BetA induced PT-pore dependent apoptosis is not cell type dependent
Because Jurkat cells are very sensitive to CsA at higher concentrations (Fig. 7) it is difficult to assess if CsA could also provide complete protection to BetA-induced apoptosis in cells without Bcl-2 over-expression. In order to test this and to generalize our findings we used A549 (lung cancer cell line) and SW480 (colon cancer cell line), which tolerate higher concentrations of CsA (up to 50 μg/ml). Strikingly, in these cell lines we clearly observed that CsA completely blocked BetA induced apoptosis, even at high concentrations (15 μg/ml) of BetA (Fig. 7). Similar results were obtained using HCT116 cells lacking Bax and Bak (Fig. 7); again CsA provided very effective protection. This indicates that the mechanism of BetA induced apoptosis depends on the PT pore and is broadly applicable in a variety of tumor cells.

BetA induced PT-pore dependent apoptosis is not cell type dependent. Jurkat wildtype, A549 (lung cancer cell line), SW480 and HCT116 Bax/Bak DKO (colon cancer cell lines) were pre-treated for 1 h with increasing concentrations of CsA and subjected to 10 or 15 μg/ml BetA for 48 h after which DNA fragmentation was assessed
Discussion
BetA is a very potent compound that is capable of killing a plethora of tumor cells. Here we demonstrate that BetA induces apoptosis in a manner that is dependent on the apoptosome, but is not affected by a lack of pro-apoptotic Bcl-2 family members Bax and Bak. BetA appears to target the mitochondrial PT pore directly and mitochondrial depolarization indeed is shown to be prevented by CsA. The anti-apoptotic Bcl-2 family members provide some protection, but this is at best limited and overcome at later time points. Multiple tumor cell lines have been shown to resist classical mitochondrial death pathways as they have a disturbed ratio of pro- and anti-apoptotic Bcl-2 family members [15]. Our report now indicates that such disturbances are to a large extent irrelevant for the apoptosis induced by BetA.
Previous studies using the neuroblastoma cell line SHEP have demonstrated BetA-induced apoptosis to be completely abrogated by Bcl-2 over-expression [12, 13], which is in apparent contrast with our data. However, the same group showed that over-expression of Bcl-2 in Jurkat cells only provided about 65% protection after 24 h treatment [2]. Similarly, it has been shown in two human glioma cell lines and in melanoma cells that over-expression of Bcl-2 only partially reduced caspase-3 activity and cell death after BetA-treatment [11, 20]. This observation is now corroborated by us, but extended to later time points and other cell lines. We found that the partial protective effect is not detected at all at later time points where the sensitivity to BetA in Bcl-2 over-expressing cells and wild-type cells was similar (Fig. 2c). It is important to note that this failure to protect is not due to the level of Bcl-2 (Fig. 2b) as etoposide-induced death is blocked at all time points tested (Fig. 2a). Previously, Chintharlapalli et al. [37, 38] reported that BetA directly affects transcription factors sp1, 3, and 4 and that this would lower transcription of survivin as well as Bcl-2. As such, BetA could lower Bcl-2 expression and thereby induce apoptosis. Although this is likely to affect sensitivity of tumor cells indirectly, we do not believe that this is the explanation for the differential sensitivity that we observe between BetA and etoposide. First and most important, Bcl-2 and Bcl-XL in our system are not dependent on their endogenous promoter, but on the strong CMV or EBV promoter and are thus not affected by sp1, 3, 4 down regulation. Secondly, even in the non-over-expressing Jurkat lines, we do not observe a decrease in Bcl-2 or Bcl-XL expression 24 h after BetA treatment.
The Bcl-2 independency of our system is even further supported by our observation that BetA-induced apoptosis is independent of the pro-apoptotic Bcl-2 family members Bax and Bak. In Bax/Bak DKO MEFs caspase-3 and PARP processing was induced with equal efficiency as in wild-type cells (Fig. 4d). Previously, Liby et al. [39] showed that Bax/Bak DKO MEFs were sensitive to BetA derivatives, but resisted BetA induced apoptosis. It is difficult to provide a rational for this discrepancy, especially as we also observe BetA-induced apoptosis in Bax/Bak deficient HCT-116 cells. We do observe a slight difference in cytochrome c release though, which is less pronounced in Bax/Bak DKO MEFs (Fig. 5a), but importantly, the amount of cells releasing cytochrome c is identical (Fig. 5b). The small difference in cytochrome c release is therefore either the result of less mitochondria per cell that release cytochrome c or that less cytochrome c is released per mitochondria. We believe this signifies the absence of an amplification loop initiated by active caspase-3, which cleaves Bid and thereby targets the mitochondria to induce maximal release. In agreement, caspase-8 processing occurs as an event downstream of the mitochondria in BetA treated neuroblastoma cells [12], and both caspase-8 and Bid-cleavage were shown for BetA treated Jurkat cells [2]. Bcl-2 over-expression and Bax/Bak deficiency could therefore disallow this amplification and cells would thereby show less cytochrome c release.
Mitochondrial membrane depolarization is a heavily debated issue in apoptosis signaling. There is agreement on its occurrence, but the causal role for cytochrome c release is clearly disputed [32]. We show that etoposide-induced loss of TMRE is blocked by Bcl-2 (Fig. 6d), and that BetA-induced depolarization is only partially prevented by Bcl-2 (Fig. 6c) but appears to serve a causal role as suggested by the inhibition of apoptosis by CsA (Fig. 6f). These observations point to a model in which BetA directly targets the pore and thereby allows cytochrome c release and downstream caspase activation. Bcl-2 family members have been shown to directly bind VDAC and regulate its pore forming activity. The pro-apoptotic member Bax promotes opening, while the anti-apoptotic members induce closure [40]. A direct Bcl-2 family member-independent opening of the PT pore by BetA is consistent with our current observations. These would be independent of Bax and Bak, but could be hampered by Bcl-2 or Bcl-XL, which independently promote closure of the pore [41]. The fact that CsA prevents pore opening and reduces apoptosis significantly, combined with the observation that this effect is complete in Bcl-2 over-expressing Jurkat cells adds to this hypothesis. It is important to note though that higher concentrations of CsA are capable of complete protection against BetA-induced apoptosis in three separate tumor lines. In Jurkat cells we failed to obtain full protection due to the high toxicity of CsA itself on these cells. However, A549 lung carcinoma cells, SW480 colon carcinoma cells as well as Bax/Bak deficient HCT-116 colon carcinoma cells all withstood CsA up to concentrations of 50 μg/ml and showed complete inhibition of BetA-induced apoptosis by CsA. Previous observations have shown that bongkrekic acid, a separate PT pore inhibitor, can also prevent BetA-induced cytotoxic effects [13] and thus support this model even further. We therefore believe that BetA targets the PT pore directly and thereby induces mitochondrial-dependent apoptosis. Other compounds, such as gossypol [42], A23187/ArA [43], and chelerythrine [44] have recently also been shown to induce Bax/Bak-independent cytochrome c release. Although for A23187/ArA-induced release this is suggested to be a serine protease dependent mechanism, chelerythrine, which shares some structural similarity with BetA, can induce release from isolated mitochondria. Although chelerythrine was developed as a Bcl-XL inhibitor, it also appears to induce mitochondrial permeabilization and apoptosis in Bax/Bak DKO cells, which is prevented by CsA [44]. Combined, this may point to a mechanism in which the lipophilic characteristics of these compounds provide them with the means to target the outer mitochondrial membrane and thereby potentially affect the PT pore. In agreement with this idea is the observation that the PT pore opening is affected by the cholesterol content of the outer mitochondrial membrane [32]. As cholesterol and BetA are structurally related, BetA could either modify this effect or have similar activity itself. Importantly, some tumor cells have been shown to contain higher cholesterol levels in their mitochondrial membrane and lowering this sensitizes them to apoptosis [32]. More importantly, reconstituted PT pores in artificial membranes localize to cholesterol areas and can be opened using long chain fatty acids [45], indicating that lipids have a strong modulating effect on the pore.
Although these data provide an appealing explanation for the broad effects of BetA on tumor cells, it still remains difficult to understand why it is without effect on untransformed cells. As mentioned, the lipid content of the mitochondria in tumor cells may be different to some extent, but this is unlikely to explain the selectivity for tumor cells. Especially as the changes observed in tumor cells rather prevent then facilitate apoptosis induction. It is possible though that another component associated with the PT-pore is a crucial determinant in the toxicity exerted by BetA. This component, hexokinase is associated with the pore on the cytoplasmic side and regulates pore opening as well as the level of glycolysis in a cell [46]. It is well established that tumor cells mainly utilize glycolysis for their ATP generation. This so called Warburg effect is potentially regulated by a differential expression level of hexokinase II in tumor versus normal cells [47]. Inactivation of hexokinase II using 3-bromopyruvate displays similar tumor selectivity as BetA [46].
In summary, we conclude that our study provides an explanation as to why BetA is a very effective and broadly applicable anti-cancer agent, even against tumor cells which have acquired resistance to other, Bcl-2 family dependent, apoptosis inducing treatments. Future experiments will provide more mechanistic insight into the exact mechanism by which BetA modulates the PT-pore and whether the tumor selectivity involves PT pore sensitivity and/or hexokinase activity.
References
Pisha E, Chai H, Lee IS et al (1995) Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat Med 1:1046–1051. doi:10.1038/nm1095-1046
Fulda S, Jeremias I, Steiner HH et al (1999) Betulinic acid: a new cytotoxic agent against malignant brain-tumor cells. Int J Cancer 82:435–441. doi:10.1002/(SICI)1097-0215(19990730)82:3<435::AID-IJC18>3.0.CO;2-1
Ehrhardt H, Fulda S, Fuhrer M et al (2004) Betulinic acid-induced apoptosis in leukemia cells. Leukemia 18:1406–1412. doi:10.1038/sj.leu.2403406
Eiznhamer DA, Xu ZQ (2004) Betulinic acid: a promising anticancer candidate. IDrugs 7:359–373
Kessler JH, Mullauer FB, de Roo GM et al (2007) Broad in vitro efficacy of plant-derived betulinic acid against cell lines derived from the most prevalent human cancer types. Cancer Lett 251:132–145. doi:10.1016/j.canlet.2006.11.003
Jung GR, Kim KJ, Choi CH et al (2007) Effect of betulinic acid on anticancer drug-resistant colon cancer cells. Basic Clin Pharmacol Toxicol 101:277–285. doi:10.1111/j.1742-7843.2007.00115.x
Zuco V, Supino R, Righetti SC et al (2002) Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett 175:17–25. doi:10.1016/S0304-3835(01)00718-2
Selzer E, Pimentel E, Wacheck V et al (2000) Effects of betulinic acid alone and in combination with irradiation in human melanoma cells. J Invest Dermatol 114:935–940. doi:10.1046/j.1523-1747.2000.00972.x
Fulda S, Friesen C, Los M et al (1997) Betulinic acid triggers CD95 (APO-1/Fas)- and p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res 57:4956–4964
Wick W, Grimmel C, Wagenknecht B et al (1999) Betulinic acid-induced apoptosis in glioma cells: a sequential requirement for new protein synthesis, formation of reactive oxygen species, and caspase processing. J Pharmacol Exp Ther 289:1306–1312
Tan Y, Yu R, Pezzuto JM (2003) Betulinic acid-induced programmed cell death in human melanoma cells involves mitogen-activated protein kinase activation. Clin Cancer Res 9:2866–2875
Fulda S, Susin SA, Kroemer G et al (1998) Molecular ordering of apoptosis induced by anticancer drugs in neuroblastoma cells. Cancer Res 58:4453–4460
Fulda S, Scaffidi C, Susin SA et al (1998) Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J Biol Chem 273:33942–33948. doi:10.1074/jbc.273.51.33942
Fulda S, Debatin KM (2000) Betulinic acid induces apoptosis through a direct effect on mitochondria in neuroectodermal tumors. Med Pediatr Oncol 35:616–618. doi:10.1002/1096-911X(20001201)35:6<616::AID-MPO27>3.0.CO;2-N
Adams JM, Cory S (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26:1324–1337. doi:10.1038/sj.onc.1210220
Leist M, Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2:589–598. doi:10.1038/35085008
de Bruin EC, Meersma D, de Wilde J et al (2003) A serine protease is involved in the initiation of DNA damage-induced apoptosis. Cell Death Differ 10:1204–1212. doi:10.1038/sj.cdd.4401296
Medema JP, Scaffidi C, Krammer PH et al (1998) Bcl-x(L) acts downstream of caspase-8 activation by the CD95 death-inducing signaling complex. J Biol Chem 273:3388–3393. doi:10.1074/jbc.273.6.3388
Waterhouse NJ, Trapani JA (2003) A new quantitative assay for cytochrome c release in apoptotic cells. Cell Death Differ 10:853–855. doi:10.1038/sj.cdd.4401263
Raisova M, Hossini AM, Eberle J et al (2001) The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J Invest Dermatol 117:333–340. doi:10.1046/j.0022-202x.2001.01409.x
Kurokawa H, Nishio K, Fukumoto H et al (1999) Alteration of caspase-3 (CPP32/Yama/apopain) in wild-type MCF-7, breast cancer cells. Oncol Rep 6:33–37
Slee EA, Adrain C, Martin SJ (2001) Executioner caspase-3,-6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem 276:7320–7326. doi:10.1074/jbc.M008363200
Puthalakath H, Strasser A (2002) Keeping killers on a tight leash: transcriptional and posttranslational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ 9:505–512. doi:10.1038/sj.cdd.4400998
Chen L, Willis SN, Wei A et al (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17:393–403. doi:10.1016/j.molcel.2004.12.030
Adams JM, Cory S (2007) Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol 19:488–496. doi:10.1016/j.coi.2007.05.004
Wei MC, Zong WX, Cheng EHY et al (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727–730. doi:10.1126/science.1059108
Zhang L, Yu J, Park BH et al (2000) Role of BAX in the apoptotic response to anticancer agents. Science 290:989–992. doi:10.1126/science.290.5493.989
Hemmati PG, Guner D, Gillissen B et al (2006) Bak functionally complements for loss of Bax during p14(ARF)-induced mitochondrial apoptosis in human cancer cells. Oncogene 25:6582–6594. doi:10.1038/sj.onc.1209668
Basanez G, Zhang J, Chau BN et al (2001) Pro-apoptotic cleavage products of Bcl-x(L) form cytochrome c-conducting pores in pure lipid membranes. J Biol Chem 276:31083–31091. doi:10.1074/jbc.M103879200
Jonas EA, Hickman JA, Chachar M et al (2004) Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc Natl Acad Sci USA 101:13590–13595. doi:10.1073/pnas.0401372101
Kirsch DG, Doseff A, Chau BN et al (1999) Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem 274:21155–21161. doi:10.1074/jbc.274.30.21155
Grimm S, Brdiczka D (2007) The permeability transition pore in cell death. Apoptosis 12:841–855. doi:10.1007/s10495-007-0747-3
Newmeyer DD, Ferguson-Miller S (2003) Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112:481–490. doi:10.1016/S0092-8674(03)00116-8
Kokoszka JE, Waymire KG, Levy SE et al (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427:461–465. doi:10.1038/nature02229
Baines CP, Kaiser RA, Sheiko T et al (2007) Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9:550–555. doi:10.1038/ncb1575
Tsujimoto Y, Shimizu S (2007) Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 12:835–840. doi:10.1007/s10495-006-0525-7
Chadalapaka G, Jutooru I, Chintharlapani S et al (2008) Curcumin decreases specificity protein expression in bladder cancer cells. Cancer Res 68:5345–5354. doi:10.1158/0008-5472.CAN-07-6805
Chintharlapalli S, Papineni S, Ramaiah SK et al (2007) Betulinic acid inhibits prostate cancer growth through inhibition of specificity protein transcription factors. Cancer Res 67:2816–2823. doi:10.1158/0008-5472.CAN-06-3735
Liby K, Honda T, Williams CR et al (2007) Novel semisynthetic analogues of betulinic acid with diverse cytoprotective, antiproliferative, and proapoptotic activities. Mol Cancer Ther 6:2113–2119. doi:10.1158/1535-7163.MCT-07-0180
Shimizu S, Narita M, Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399:483–487. doi:10.1038/20959
Shimizu S, Shinohara Y, Tsujimoto Y (2000) Bax and Bcl-x(L) independently regulate apoptotic changes of yeast mitochondria that require VDAC but not adenine nucleotide translocator. Oncogene 19:4309–4318. doi:10.1038/sj.onc.1203788
Lei XB, Chen YY, Du GH et al (2006) Gossypol induces Bax/Bak-independent activation of apoptosis and cytochrome c release via a conformational change in Bcl-2. FASEB J 20:2147–2149. doi:10.1096/fj.05-5665fje
Mizuta T, Shimizu S, Matsuoka Y et al (2007) A Bax/Bak-independent mechanism of cytochrome c release. J Biol Chem 282:16623–16630. doi:10.1074/jbc.M611060200
Wan KF, Chan SL, Sukumaran SK et al (2008) Chelerythrine induces apoptosis through a Bax/Bak-independent mitochondrial mechanism. J Biol Chem 283:8423–8433. doi:10.1074/jbc.M707687200
Wieckowski MR, Brdiczka D, Wojtczak L (2000) Long-chain fatty acids promote opening of the reconstituted mitochondrial permeability transition pore. FEBS Lett 484:61–64. doi:10.1016/S0014-5793(00)02127-X
Mathupala SP, Ko YH, Pedersen PL (2006) Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25:4777–4786. doi:10.1038/sj.onc.1209603
Pedersen PL (2007) Warburg, me and hexokinase 2: multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr 39:211–222. doi:10.1007/s10863-007-9094-x
Acknowledgments
We would like to thank Jannie Borst (NKI, Amsterdam, The Netherlands), Marja Jäättelä (Danish Cancer Society, Copenhagen, Denmark), John Blenis (Harvard, Boston, USA), Stanley Korsmeyer, Tak Mak (University of Toronto, Canada), Richard Flavell (Yale, New Haven, USA), Bert Vogelstein (Johns Hopkins University) and Chinna Chinnadurai (St Louis School of Medicine) for their kind and rapid actions in providing cells that were of great value to this study and Eric Eldering (AMC, Amsterdam, The Netherlands) for anti Bak antibody. This work was supported by a grant of the Stichting Nationaal Fonds tegen Kanker (SNFK), Amsterdam, The Netherlands.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mullauer, F.B., Kessler, J.H. & Medema, J.P. Betulinic acid induces cytochrome c release and apoptosis in a Bax/Bak-independent, permeability transition pore dependent fashion. Apoptosis 14, 191–202 (2009). https://doi.org/10.1007/s10495-008-0290-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0290-x