
Abstract
Shwachman–Diamond syndrome (SDS) is an inherited disorder characterized by reduced cellularity in the bone marrow and exocrine pancreas. Most patients have mutations in the SBDS gene, whose functions are unknown. We previously showed that cells deficient in the SBDS protein are characterized by accelerated apoptosis and Fas hypersensitivity, suggesting that the protein might play an important role in Fas-mediated apoptosis. To study the mechanism of Fas hypersensitivity, we compared shRNA-mediated SBDS-knockdown HeLa cells and SDS marrow CD34+ cells for their sensitivity to several groups of apoptosis inducers. Marked hypersensitivity was noticed in response to Fas stimulation, but not to tumor necrosis factor-α, DNA-damaging agents, transcription inhibition or protein synthesis inhibition. To identify the Fas signaling factors that cause hypersensitivity, we analyzed the expression of the pathway’s proteins. We found that Fas accumulated at the plasma membrane in SBDS-knockdown cells with corresponding expression of Fas transcript 1, the main Fas transcript which contains both the transmembrane domain and the death domain. However, the total levels of Fas protein and mRNA were comparable to controls, and Fas internalization occurred normally. Expression of FADD, caspase-8 and -3 were not elevated and the pathway inhibitors: ERK, c-FLIP and XIAP were not decreased. These results suggest that SBDS loss results in abnormal accumulation of Fas at the plasma membrane, where it sensitizes the cells to stimulation by Fas ligand.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Shwachman–Diamond syndrome (SDS) is an autosomal recessive disorder characterized mainly by short stature and reduced cellularity in the bone marrow and exocrine pancreas [1, 2]. Patients with the syndrome suffer mainly from cytopenia, malabsorption and short stature. In addition, there is marked propensity for myelodysplastic syndrome and leukemia [3–5]. Recently, the gene associated with the syndrome has been identified and designated as SBDS. SBDS is located at 7q11 and highly conserved from archaea to humans. Approximately 90% of the SDS patients have mutations in SBDS. Common mutations arise from gene conversion events with its pseudogene, SBDSP, whose sequence is 97% identical to SBDS [6]. There are two common mutations 183-184TA>CT (nonsense mutation) and 258+2T>C (intronic mutation; predicted to result in alternative splicing and frameshift reading). The mutations are predicted to cause reduced expression of the protein.
The functions of the SBDS protein are unknown; however, various data suggest a role of SBDS in regulating apoptosis, [7, 8] ribosomal biogenesis [9–13] and chemotaxis [14–16]. Since SDS marrows are hypocellular, [1, 2, 17] we previously asked whether SDS marrow progenitors undergo accelerated apoptosis. We found that SDS hematopoietic progenitors exhibit higher apoptosis rates than normal cells, [7] a process which has recently been suggested to play a major pathogenetic role in cytopenia related to several bone marrow failure syndromes [18–22]. Since the Fas signaling pathway is a key regulator of apoptosis in physiologic and disease related states, [23, 24] we have tested SDS marrow progenitor cells, and found them to be hypersensitive to direct Fas stimulation [7]. In many physiological and pathological conditions, including myelodysplastic syndromes, regulation of the Fas signaling pathway has been shown to be at the Fas protein level [25, 26]. Therefore, we evaluated SDS marrow cells for Fas protein expression and found overexpression throughout maturation: CD34+, CD34−, CD34−/CD38+, CD34−/CD38−, suggesting that apoptosis starts early during hematopoiesis [7]. In contrast to Fas, the Bax/Bcl-2 and Bax/Bcl-XL expression ratios at the protein and mRNA level did not manifest pro-apoptosis expression pattern in SBDS-deficient cells [8]. Using apoptosis blockers we showed that the Fas pathway was the predominant apoptosis pathway responsible for the decreased cell growth in SBDS-deficient cells [8].
Since our previous work clearly demonstrated that SBDS-deficient cells undergo accelerated apoptosis and are hypersensitive to Fas stimulation, we investigated herein the mechanism for Fas hypersensitivity upon SBDS loss. To do so, we first asked whether the hypersensitivity of SBDS-deficient cells is specific to Fas stimulation, or occurs indiscriminately in response to a variety of apoptosis cues. To determine the proteins that confer Fas hypersensitivity in SBDS-deficient cells, we analyzed the expression of the various Fas pathway proteins and its inhibitors. Last, we focused on Fas and studied whether alterations in protein localization or mRNA expression can play a role in causing activation of the pathway.
Materials and methods
Patients and bone marrow samples
The studies were approved by the Institutional Research Ethics Board, and informed written consent was obtained from patients, controls, or their legal guardians prior to sample collection. Patients were diagnosed with SDS based on institutional clinical criteria, [27] which included clear evidence for both hematological and exocrine pancreatic dysfunction. For the various experiments, marrow samples from 12 patients were used. Nine of the patients were tested for mutations in the SBDS gene, and were found to have biallelic mutations. No patient had evidence of malignant transformation at the time of bone marrow sample collection.
Bone marrow aspirates were obtained from the posterior superior iliac crest, put into Iscove’s medium containing ten units of preservative-free heparin per milliliter, layered over Ficoll-Hipaque and centrifuged for 25 min at 1600 rpm. The light-density cell fraction was collected and washed.
Reagents
Custom chicken anti-human SBDS antibody was generated by immunization of the animals with the C-terminal of SBDS and purified by Invitrogen (Carlsbad, CA). Antibodies to Fas, FADD, and caspase-8 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to XIAP, Erk1/2, PI3K, Akt, Jak2, Stat1 and phosphorylated Stat1 and Erk1/2 were from Cell Signaling Technology (Danvers, MA). Anti c-FLIP antibody was a gift from Dr. J. C. Reed, Burham Institute. Phycoerythrin-cyanin-5 (Cy5)-conjugated anti-human Fas and FITC-conjugated goat-anti-mouse IgM was from BD Pharmingen (Franklin Lakes, NJ). The agonistic anti-Fas IgM antibody (CH-11) and blocking anti-Fas antibody (cZB4) were purchased from Immunotech (Marseille, France). Fas ligand was purchased from R&D Systems (Minneapolis, MN). Actinomycin D, α-amanitin, cycloheximde, cisplatin and doxorubicin were purchased from Sigma–Aldrich (St Louis, MO). Etoposide was purchased from Calbiochem (San-Diego, CA). TNF-α was from R&D Systems (Minneapolis, MN), and interferon-γ was from InterMune Pharmaceuticals, Inc. (Palo Alto, CA). The Caspase 8 inhibitor (Z-IETD-FMK) was purchased from Trevigen, Gaithersburg, MD.
Cell lines
HeLa human cervical cancer cells were maintained in Dulbecco Modified Eagle’s Medium supplemented with 10% fetal bovine serum at 37°C in 5% CO2.
Short hairpin RNA (shRNA)-mediated HeLa knockdown cells were generated as previously established [8]. In short, three different shRNA expression cassettes containing either SBDS mRNA sequences (shSBDS-1 & shSBDS-3) or scrambled sequence control (shSCR) were synthesized downstream to a U6 promotor. Cassettes were then cloned into a pSEC/Neo plasmid (Ambion, Austin, TX) and transfected into HeLa cells using Lipofectamine 2000 (Invitrogen). After 72 h, cells were selected with G418 (Invitrogen). According to the shRNA expression construct, we termed the lines HeLa/shSBDS-1, HeLa/shSBDS-3 and HeLa/shSCR. SBDS-knockdown was confirmed by Western blotting using a chicken anti-SBDS antibody (Supplemental Data, Fig. 1). We chose HeLa cells as a model, because they have been used extensively to study protein function by shRNA, [28] they express Fas and can induce to undergo apoptosis [29, 30].

Sensitivity of SBDS-knockdown cells to Fas stimulation. a SBDS-knockdown cells were either not treated or treated with CH-11 (0.02 μg/ml) for 48h. The cell morphology was then evaluated under the microscope. b SBDS-knockdown cells were treated with various concentrations of activating anti-Fas antibody, CH11 or Fas ligand (c) for 48 h and then analyzed by MTT assay. The means results (±SD) of three replicates relative to the values obtained from the same cell type without treatment are presented. d Nuclear staining of Hela-shSBDS-3 using Hoechst33342 with or without Fas stimulation. (WT, HeLa/WT; Scr, HeLa/shSCR; S-1, HeLa/shSBDS-1; S-3, HeLa/shSBDS-3)
Methyl–thiazol–tetrazolium (MTT) assay
Cells were plated into 96-well plates in triplicate wells (4 × 103 cells/well) and incubated overnight at 37°C before treated with various apoptosis inducers. After 48 h the cytotoxic activity was measured by the reduction in the conversion rate of the MTT dye (3-4,5-dimethylthiazol-2-yl-2, 5-diphenyltetrazolium bromide) (ATCC, Manassas, VA, USA) to a blue–black formazan product. Absorbance at 570 nm was measured in a micro plate reader (Bio-Tek Instruments, Inc., Winooski, VT, USA).
Flow cytometry
Double staining for annexin V and propidium iodide was done as previously described [7] with minor modifications. Briefly, to a 5 × 105 cell mixture, FITC-conjugated annexin V (1 μL, R&D Systems, Minneapolis, MN) and propidium iodide (10 μL, R&D Systems) were added, and cells were incubated at room temperature in the dark for 15 min. Subsequently, binding buffer (R&D Systems) was added, and cells were immediately analyzed by Coulter Epics XL-MCL flow cytometer (Coulter, Hialeah, FL).
Determination of apoptotic events by DNA content analysis was performed as previously described [8]. In brief, 5 × 105 cells were washed with phosphate-buffered saline, fixed with 70% ethanol, re-washed and incubated at 4°C for additional 1 h with propidium iodide, RNaseA and triton-×-100. DNA content was then analyzed by flow cytometry and sub-G1 cell population, which indicates apoptotic cells, was determined.
For evaluation of plasma membrane Fas expression, 1 × 106 cell mixture were incubated with either 2 μg/ml of Cy5- conjugated anti-Fas antibody or isotype IgG1 in the dark for 30 min on ice and analyzed by flow cytometry as previously described [7].
Hematopoietic cell enrichment
Light-density marrow mononuclear cells obtained after Ficoll-Hipaque separation underwent CD34+ or CD33+ cell enrichment by the Mini-MACS immunomagnetic separation system (Miltenyi Biotec, Auburn, CA) as previously described [7].
Clonogenic assays
Marrow CD34+ cells were plated in duplicates at a density of 1 × 103 cells/1 ml dish with serum free medium (MethoCult SF 4436 from Stem Cell Technologies, Vancouver, Canada) containing methylcellulose, Iscove’s medium, bovine serum albumin, 2-mercaptoethanol, L-glutamine, insulin, human transferrin, stem cell factor, granulocyte macrophage-colony stimulating factor, interleukin-3, interleukin-6, granulocyte-colony stimulating factor and erythropoietin. Additional duplicate cultures were plated in the same conditions, but with escalating concentrations of CH11, etoposide, α-amanitin or cycloheximide, or were treated with γ-radiation 24 h after plating. Cultures were incubated at 37°C in a humidified atmosphere (5% CO2 and air). Colonies of 50 cells or more were scored after 14 days under an inverted microscope.
Morphology
Cells were plated into 96-well plates in triplicate wells (4 × 103 cells/well) and incubated overnight at 37°C. After 48 h of treatment with 0.02 μg/ml of CH-11, the morphology of the cells were examined and imaged under an inverted light microscope. For nuclear staining, cells were incubated on coverslips, and treated with 0.02 μg/ml of CH-11. After 48 h, cells were washed with phosphate-buffered saline, fixed with 4% paraformaldehyde, and then stained with Hoechst 33342 (Molecular Probes Inc., Carlsbad, CA). The stained cells were examined using fluorescence microscopy (Carl Zeiss, Gottingen, Germany).
Confocal microscopy
To study whether SBDS-knockdown affects the subcellular localization of Fas, cells were plated on coverslips for 24 h. Cells were then washed and incubated with 2 μg/ml PE-Cy5-conjugated anti-Fas antibody for 30 min on ice. After washing, cells were fixed with 4% cold paraformaldehyde and observed under confocal microscopy. The fluorescent signal at the plasma membrane was identified after merging the figures obtained by the fluorescent filter with that from differential interference contrast, and the fluorescent intensity in cells from 4 to 5 slides from three separate experiments were quantified using ImageJ Software (US National Institutes of Health, Bethesda, MD).
Western blotting
Cultures were washed once, and cells were scraped in phosphate-buffered saline. After centrifugation, pellets were incubated with radioimmunoprecipitation assay buffer on ice for 45 min and centrifuged at 15,000 rpm for 15 min. The supernatant was collected and protein concentration was measured using BioRad protein assay solution. Equal amounts of cell lysate were loaded onto a 10–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred to a polyvinylidene difluoride or nitrocellulose membrane. Membranes were rinsed in Tris-buffered saline (TBS) and incubated in blocking solution (5% non-fat dry milk in TBS-Tween 20) at room temperature for 1 h. Blocked membranes were incubated with a primary antibody at 4°C overnight and washed, followed by incubation with a secondary antibody at room temperature for 1.5 h. After washing, the proteins were visualized using enhanced chemiluminescence (GE, Healthcare).
Fas protein expression was also quantified by a fluorescent detection system. The experimental procedure was similar to the standard western blotting described above except for using an infrared dye 680 Goat anti-mouse IgG (Li-Cor Biosciences, Lincoln, Nebraska) as a secondary antibody for a mouse anti-human Fas antibody, and infrared dye 800 goat anti-rabbit IgG (Li-Cor Biosciences) for a rabbit anti-human actin. The proteins on the membrane were visualized and quantified using the Odyssey® Infrared Imaging System (Li-Cor Biosciences).
Receptor internalization
HeLa cells (1 × 106) were incubated with 2 μg/ml of the agonist anti-Fas IgM antibody CH-11 for 30 min on ice. Unbound antibody was removed by washing and cells were incubated with Dulbecco Modified Eagle’s Medium/10% fetal bovine serum either at 37°C to induce internalization or at 4°C for 1h. After washing, cells were incubated with 2 μg/ml Fluorescein isothiocyanate (FITC)-conjugated goat-anti-mouse IgM for 30 min on ice. The cells were then washed, and Fas expression at the plasma membrane was quantified by flow cytometry. The internalized Fas was defined as the ratio of peak Fas expression at 37°C to its expression at 4°C as previously described [26].
Quantitative real time polymerase chain reaction (PCR)
To study whether SBDS-deficiency leads to upregulation of Fas at the mRNA level we measured Fas levels in SDS cells vs. healthy controls and in SBDS-knockdown cells and compared the levels to those from HeLa/WT and HeLa/shSCR control cells. Quantitative real-time PCR using SYBR green technology was used as previously described [31]. Total RNA was used to synthesize complementary DNA (cDNA) using the Advantage RT-for-PCR kit from Clontech (Palo Alto, USA) according to the manufacturer’s instructions. The primers used for amplification of the FAS gene were: forward primer: 5′CTCCTACCTCTGGTTCTT-3′ at exon 1 and 5′-TGTCAGTCACTTGGGCATT-3′ at exon 2. The primers used for the β-actin control gene were: forward primer: 5′-AGCCTCGCCTTTGCCGA-3′ at exon 5, and backward primer 5′-CTGGTGCCTGGGGCG-3′ at exon 6. cDNA and primers were added to a SYBR green Master Mix (PE Applied Biosystems, Foster City, CA). Two steps PCR thermal cycling for DNA amplification and real time data acquisition were performed with an ABI PRISM 7700 Sequence Detection System using the following cycle conditions: 50°C for 2 min × 1 cycle, 95°C for 10 min × 1 cycle, and 95°C for 15 s followed by 60°C for 1 min × 45 cycles. Each cDNA sample was assayed in triplicates. A non-template control and a non-primer control were cycled in parallel each run. Baseline and threshold values were set, and fluorescence data were analyzed and expressed as CT, namely the number of cycles needed to generate a fluorescent signal above a predefined threshold.
Multiplex PCR
Fas transcript 1 is the maim transcript involved in transmission the death signal from Fas ligand. Herein we asked whether the accumulation of Fas at the plasma membrane level and transmission of the death signal in SBDS-deficient cells is associated with the expression of this transcript. Since Fas has multiple alternatively-spliced variants we were not able to identify primers that amplify exclusively transcripts 1 for real-time PCR. Therefore, we used a primer set that can amplify fragments of different sizes from the transcripts 1 and 2 by multiplex PCR and can be identified using an agarose gel. Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA) as previously described [8]. RNA underwent reverse transcription using the Advantage RT-for-PCR kit (Clontech, Palo Alto, USA) and oligo dT primers according to the manufacturer’s instructions. cDNA product aliquots underwent multiplex PCR of the Fas and G3APDH genes. For the Fas gene the forward primer 5′- GAAGGACATGGCTTAGAAGTGG -3′ (on exon 3,4) and the reverse primer 5′- GCCACTGTTTCAGGATTTAAGG -3′ (on exon 7,8) were used, with a predicted PCR product of 338 bp. For the G3PDH gene the forward primer 5′- TGAAGGTCGGAGTCAACGGATTTGGT -3′ and the reverse primer 5′- CATGTGGGCCATGAGGTCCACCAC -3′ were used, with a predicted PCR product of 983 bp. An aliquot of the completed PCR reaction was fractionated on 1% agarose gel by electrophoresis, autoradiographed, and scanned by the laser scanning densitometer (SynGene, Frederick, MD).
Results
Establishment of SBDS-knockdown cell lines using shRNA
To study the role of SBDS in apoptosis, we knocked down the gene in HeLa cells using shRNA. We generated three cell lines: HeLa/shSBDS-1 and HeLa/shSBDS-3, as well as a control cell line expressing scrambled RNA cassette, HeLa/shSCR. SBDS knockdown was confirmed by Western blotting using an anti-SBDS antibody (Supplemental Data, Fig. 1), The control cell line, HeLa/shSCR, had the same level of SBDS expression as the wild type cells. The SBDS levels mimic the levels in cells from SDS patients [8, 9]. Despite a clear effect of knocking down SBDS on cell survival (see below), this effect was not absolute, and the clones could be maintained for at least 3 months, cryopreserved, thawed and re-cultured.
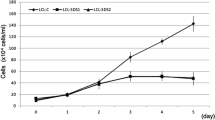
As we have previously shown, [8] SBDS-knockdown resulted in increased apoptotic cell death. Exponentially growing SBDS-knockdown cells showed 24% sub-G1 cell population for HeLa-shSBDS-1 and 33% for HeLa/shSBDS-3 compared to 8% for HeLa/WT and 12% for HeLa/shSCR cells (Data not shown). Similarly, morphological examination of the cells under a light microscope showed prominence of small and round or floating cells (Fig. 1a).
Sensitivity of the SBDS-knockdown cells to apoptosis inducers
To further characterize the effect of SBDS-knockdown on apoptosis, we studied the sensitivity of HeLa/shSBDS-1 and -3, HeLa/shSCR and HeLa/WT to four groups of apoptosis inducers. Dose-response curves were obtained by MTT assay 48 h after treating the cells with the reagents.
We used two different Fas stimulators to study whether SBDS-knockdown results in hypersensitivity to Fas stimulation: CH11, which is an activating anti-Fas antibody and Fas ligand, which is the physiological stimulator of Fas. Interestingly, SBDS-knockdown cells were markedly hypersensitive to both Fas stimulators. For example, while 0.1 μg/ml of CH-11 reduced the survival fraction to 50% in wild-type and control cells, a similar effect was obtained with only 0.01 μg/ml in the SBDS–knockdown cells (Fig. 1b). Similar prominent hypersensitivity was observed when Fas ligand was used (Fig. 1c). When the cells were examined under the microscope 48 h after treatment with CH-11, most SBDS-knockdown cells were dying, while the wild type and control cells were still mostly intact (Fig. 2a). Staining the cells with Hoechst 33342 showed the typical feature of apoptosis such as chromatin condensation and nuclear fragmentation (Fig. 1d). To study whether the Fas hypersensitivity of SBDS-knockdown cells is due to apoptosis, we evaluated annexin V binding after rescue experiments with a blocking anti-Fas antibody and a caspase 8 inhibitor. Indeed, blocking the Fas pathway by either a caspase 8 inhibitor or a blocking anti-Fas antibody prominently reduced spontaneous and CH11-induced apoptosis (Supplemental Data Fig. 2).

Sensitivity of SBDS-knockdown cells to various apoptosis agents: SBDS-knockdown cells were treated with various concentrations of the apoptosis inducers for 48 hours and then analyzed for cell viability by a MTT assay. The means (±SD) of al least 3 replicates are presented. The results were normalized to blank wells and expressed relative to the values obtained from the same cell type without treatment. The apoptosis inducers included TNF-α (a), Radiation (b), Cisplatinum (c), Etoposide (d), Doxorubicin (e), Actinomycin (f), α-Amanitin (g), Cycloheximide (h). (WT, HeLa/WT; Scr, HeLa/shSCR; S-1, HeLa/shSBDS-1; S-3, HeLa/shSBDS-3)
To determine the molecular events that lead to Fas hypersensitivity, we took advantage of the fact that stimulations of both TNF-α and Fas activate several common downstream proteins, including FADD, caspase-8 and caspase-3. We therefore, tested whether SBDS-knockdown cells are hypersensitive also to TNF-α. Importantly, there were no differences between the SBDS-knockdown cells and the HeLa/shSCR and HeLa/WT control cells in their sensitivity to TNF-α stimulation (Fig. 2a), suggesting that the hypersensitivity to Fas stimulation resides at the receptor level.
It has been debated whether patients with SDS are more sensitive to standard chemotherapy and radiation than patients with no inherited blood dyscrasias [32–34]. Interestingly, SBDS-knockdown cells were not hypersensitive to the genotoxic agents: γ-radiation, cisplatinum, etoposide and doxorubicin (Fig. 2b–3e). Since SBDS has been postulated to be involved in RNA processing and ribosome biogenesis, we studied the sensitivity of SBDS-knockdown cells to transcription inhibition by actinomycin D and α-amanitin. In contrast to Fas stimulation, the sensitivity to the transcription inhibitors was not remarkably different among the four cell lines (Fig. 2f, g). Similarly, the sensitivity to the protein synthesis blocker, cycloheximide, was not different between the SBDS-deficient and control cells (Fig. 2h).

Sensitivity of hematopoietic progenitor cells to various apoptosis inducers: Marrow CD34+ cells were plated in clonogenic assay with or without various apoptosis inducers at escalating concentrations. After 14 days the total numbers of colonies were determined under inverted microscope. The apoptosis inducers included CH11 (a), γ-radiation (b), etoposide (c), Cycloheximide (d), α-amanitine (e). f Two million freshly obtained marrow mononuclear cells from SDS patients (n = 11) and healthy subjects (n = 3) were plated in Iscove’s medium with 10% fetal calf serum. Duplicate cultures were irradiated with 15 gray immediately after plating. The cells were then incubated for 24 h and analyzed for apoptotic events by annexin V/propidium iodide staining. The right panel shows the percentages of apoptotic cells in the cultured marrow mononuclear cells, and the left panel show the fold increase in apoptosis cells after irradiation
Growth potential of SBDS-/- marrow CD34+ cells in the presence of apoptosis inducers
To study whether the prominent hypersensitivity to Fas stimulation is confined to SBDS-knockdown HeLa cells or occurs also in primary SDS patients’ hematopoietic progenitor cells, we tested CD34+ cell from two patients who are double heterozygous for the 183-184TA>CT+258+2T>C/258+2T>C mutations and one patient who is double heterozygous for the 183-184TA>CT/258+2T>C mutations. Western blotting of lymphoblasts from one patient from each group showed no detectable SBDS expression (Data not shown). Due to the rareness of the diseases and the paucity of CD34+ cells in marrows from these patients, [17] we were able to test only one–two patients for each apoptosis inducer in duplicate cultures. To overcome this problem, we performed dose response in each individual sample. Indeed, very important observations have been made, which strengthened the results from the SBDS-knockdown cells.
We have previously shown that incubation of marrow CD34+ cells from SDS patients (n = 6) in serum-based clonogenic assay containing 2 μg/ml CH11 resulted in 75% reduction in colony formation compared to 40% in normal controls (n = 4) (P < 0.05) [9]. Prominent differences between SBDS-/- cells and controls were also seen in the present study using serum-free methylcelluose culture at the same concentration of CH11 (Fig. 3a). The total number of colonies in the SDS culture after stimulation with 2 μg/ml of CH11 was 54% lower than the colony numbers where no CH11 was added. The reduction of the colony numbers in the CH11-treated culture with normal control cells was only by 28%. In contrast, treatment of the cells with γ-radiation (Fig. 3b), etoposide (Fig. 3c) and cycloheximide (Fig. 3d) did not result in any differences in colony formation between SBDS−/− cells and controls. Treatment of the cells with α-amanitin showed substantial overlapped results between patients and controls, however, a trend towards lower colony formation was seen with the lower concentration (0.5 μg/ml) and not with the higher concentration (1 μg/ml) (Fig. 3e). The mean patients’ colony numbers was reduced by 37% (range: 16–57%), compared to 13% (range: −10–39%) of healthy subject. Larger numbers of patients are needed to decipher whether SBDS-deficient cells have certain degree of hypersensitivity also to low dose α-amanitin.
For sensitivity to DNA damaging agents, we also plated 2 million freshly obtained marrow mononuclear cells after Ficoll separation in suspension cultures containing Iscove’s medium and 10% fetal calf serum. Duplicate cultures were treated with 15 gray γ-irradiation immediately after plating. After incubation for 24 h, non-adherent cells were harvested and stained with annexin V/propidium iodide and analyzed by flow cytometry (Fig. 3f). The mean baseline apoptosis rates as determined by annexin V+/propidium iodide-events were higher in the patients (20%, n = 11) compared to controls (10%, n = 3) (P = 0.04). Interestingly, the fold increase in apoptotic cells among the SDS patients cells after γ-radiation was only 1.5 vs. 2.3 among the normal samples (P = 0.08).
Fas protein expression and subcellular localization
The results above demonstrate prominent hypersensitivity of primary and shRNA-mediated knockdown SBDS-deficient cells to Fas stimulation. To identify the factors in the Fas signaling pathway that are responsible for this hypersensitivity, we analyzed the expression of the Fas signaling pathway-related proteins. Since changes in Fas protein expression have been reported to affect the apoptosis response to Fas stimulation, we first studied the expression of Fas protein at the plasma membrane by flow cytometry. Fas expression in the HeLa/shSBDS-1 and -3 was 1.8 and 2.6 fold higher, respectively, than the HeLa/shSCR control (Fig. 4a). This is congruent with our previous observations in SDS marrow progenitor cells [7].

Fas protein expression in SBDS-knockdown. a Cells were stained with Cy5-conjugated anti-Fas antibody and assayed by flow cytometry for Fas plasma membrane expression. Two separate sets of experiments are shown. b Fas protein expression was assayed by standard western blotting and detection by chemiluminecence. The ratios of Fas expression to the β-actin internal control are shown beneath the respective bands. c Fas protein expression by western blotting using an infrared detection system. The red dye stains for Fas and the green dye stains for β-actin. (WT, HeLa/WT; Scr, HeLa/shSCR; S-1, HeLa/shSBDS-1; S-3, HeLa/shSBDS-3)
To determine whether Fas overexpression at the plasma membrane levels is due to increased total intracellular protein levels we evaluated the total Fas protein levels by Western blotting using standard horseradish peroxidase (HRP)-conjugated and more accurately by infrared-conjugated antibodies. We used a polyclonal anti-Fas antibody from Santa Cruz (C-20) that recognizes the last 50 amino acids at the C-terminus of the protein. In both methods, the total Fas protein (a band at approximately 39kD) was similar in the SBDS-knockdown and the control HeLa/WT and HeLa/shSCR cells (Fig. 4b, c). These data suggest that the Fas cell surface overexpression after SBDS loss is not due to an increase in its total cellular levels.
Since the plasma membrane Fas expression but not the total Fas levels differ between SBDS-knockdown and control cells, we next evaluated whether SBDS loss affects the subcellular localization of Fas by confocal microscopy. We found that Fas accumulated at the plasma membrane level in the SBDS-knockdown cells, while the distribution was more diffuse in the control cells (Fig. 5a). To quantitate the difference in Fas accumulation at the plasma membrane in the SBDS-knockdown cells, we measured the relative fluorescent signal intensity of Fas in the various cell lines. The means of the fluorescent intensity units in the HeLa/shSBDS-1 cells and the HeLa/shSBDS-S3 cells were significantly lower than the controls (Fig. 5b). Similarly the ratio of plasma membrane Fas expression vs. cytoplasmic Fas was significantly higher in the SBDS-knockdown cells than the controls (Fig. 5c).

Fas subcellular localization. a SBDS-knockdown and control cells were stained with Cy-5-conjugated anti-Fas antibody (left panel). Cells were co-stained with FITC-conjugated cholera toxin B (CT-B) for cell membrane marking (middle panel). The right panel shows merged images of both fluorescents. b The fluorescent signal intensity of plasma membrane Fas relative to CT-B was analyzed by the Velocity software (Improvision Inc.). The mean (± SEM) of the signal intensity in the knockdown cells and the controls is shown. c The ratio of Cy-5 fluorescent intensity in the plasma membrane to cytoplasm is compared between the various cell lines. (WT, HeLa/WT; Scr, HeLa/shSCR; S-1, HeLa/shSBDS-1; S-3, HeLa/shSBDS-3)
Expression of other Fas-signaling related proteins in SBDS-knockdown cells
Stimulation of Fas triggers recruitment of FADD and procaspase-8 to the receptor. This results in a cleavage and activation of caspase-8 and subsequently of caspase-3, and leads to other biochemical changes associated with apoptosis such as DNA fragmentation. It has been reported that the expression of FADD and procaspase-8 can affect the cellular sensitivity to Fas stimulation [35]. Thus, we examined the expression of these molecules before and 6 h after Fas stimulation. Western blotting analysis showed no significant increase in the protein level of FADD, procaspase-8 and procaspase-3 (Fig. 6). As expected from the hypersensitivity experiments to Fas, cleavage of caspase-8 in response to Fas stimulation was prominent in the SBDS-knockdown cells (Fig. 6).

Western blotting of Fas signaling-related proteins in the SBDS-knockdown and control cells before (−) and 6 h after (+) treatment with CH-11 0.02 μg/ml. (WT, HeLa/WT; Scr, HeLa/shSCR; S-1, HeLa/shSBDS-1; S-3, HeLa/shSBDS-3)
Next, we studied whether the Fas hypersensitivity of SBDS-knockdown cells is due to low expression of proteins, which suppress Fas-mediated apoptosis: e.g., XIAP, c-FLIPL, c-FLIPS and Erk. However, there was no substantial decrease in the levels of these inhibitors in the SBDS-knockdown cells compared to the control cells (Data not shown).
Accumulation of Fas at the plasma membrane is not associated with a defect in its internalization
Decreased Fas internalization can lead to increased Fas accumulation at the plasma membrane level. Although Fas internalization is required for Fas signaling, hypersensitivity to Fas stimulation was described when Fas internalization was partially impaired and led to sustained availability of Fas to bind Fas ligand [26]. We thus studied whether Fas internalization is impaired in the SBDS-knockdown cells. We used standard Fas activation with CH11 followed by incubation in culture medium at warm temperature to induce internalization of the receptor. The rates of Fas internalization in HeLa/shSBDS-1 (80%) and in HeLa/shSBDS-3 (70%) cells were not decreased compared to HeLa/WT (50%) and HeLa/shSCR (50%) cells (Fig. 7). In fact, there was slight increase in Fas internalization in the SBDS-knockdown cells, which may reflect the increase activation of the receptor.

Fas internalization. SBDS-knockdown and control HeLa cells were incubated with 2 μg/ml of CH11 antibody followed by either incubation at 37°C to induce internalization or at 4°C (no internalization control). The cells were then analyzed for Fas cell surface expression by flow cytometry. The mean internalization rates of two separate sets of experiments are depicted at the bottom of each graph. Mean internalization rates were defined as the ratio of the peak Fas expression at 37°C to the peak Fas expression at 4°C × 100. The shaded areas represent Fas expression at 4°C, and the open areas represent its expression after incubation at 37°C. (WT, HeLa/WT; Scr, HeLa/shSCR; S-3, HeLa/shSBDS-3)
Expression of Fas-trafficking related proteins
Since it has been reported that interferon-γ induces Fas trafficking to cell surface through activation of PI3K and Akt or Jak2 and Stat1 in vascular smooth muscle cells [46], we further examined the expression of these proteins in SBDS-knockdown and control cells. However, the expression of all the proteins was not increased in SBDS-knockdown cells in comparison to the control cells (Supplemental Data Fig. 3 and data not shown), suggesting that these proteins may not play a role in cell surface accumulation of Fas in SBDS-deficient cells. Similarly, expression of dynamin 1/2 and FAP1, which are also involved in Fas trafficking, were comparable between the SBDS-knockdown and control cells (Data not shown).
Fas mRNA expression
Since total Fas protein levels in SBDS-knockdown cells were similar to controls, we anticipated finding no dysregulation of Fas at the mRNA levels. Indeed, total Fas mRNA levels as determined by real-time PCR in the SBDS-knockdown cells (Fig. 8a) and marrow cells from SDS patients (Fig. 8b) were similar to those of controls. Next, we asked whether SBDS-deficient cells expressed a functional Fas transcript and what its expression level was. We studied the transcripts that are expressed in the SBDS-knockdown cells by amplification of Fas using primers that flank the entire open reading frame and cloned the PCR products into pCR 2.1-TOPO vector (Invitrogen). After propagation into DH-α Escherichia coli cells, we analyzed 20 clones from each cell types by amplification and digestion with BamHI. We found similar types of transcripts in the SBDS-knockdown and controls cells, and the major transcript in all cell types was transcript 1 (Data not shown). To quantify the relative expression of transcript 1, we performed multiplex PCR. The primer set amplifies transcript 1 and 2 (Supplemental Data, Fig. 2), which can be recognized on an agarose gel. The results were normalized to G3PDH as an internal control. The expression levels of transcript 1 were approximately 60% higher in the HeLa/shSBDS-3 (Fig. 8c) and in SDS marrow CD33+ cells (Fig. 8d) compared to the controls. One of the healthy controls had a prominent expression of transcript 2, which does not contain the transmembrane domain (Fig. 8d, lane 1).

Fas mRNA expression. a RNA from untreated exponentially growing SBDS-knockdown and control cells was extracted and analyzed for Fas mRNA expression by real-time PCR. The β-actin (ACTB) gene was used as an internal control. b Fas mRNA expression was analyzed in RNA samples from marrow mononuclear cells by real-time PCR. c Cellular RNA after reverse transcription was analyzed by multiplex PCR using primers that can amplify transcript one and two. Only transcript one could be detected in this experiment. The ratios of Fas expression to the G3PDH internal control are shown beneath the respective bands. d Fas transcript one and G3PDH expression in the marrow CD33+ cells from SDS patients and controls was analyzed by multiplex PCR as described in c For marrow CD33+ cells a different set of G3PDH was necessary to successfully amplify both FAS and G3PDH. The ratios of Fas expression to the G3PDH internal control are shown beneath the respective bands. (WT, HeLa/WT; Scr, HeLa/shSCR; S-1, HeLa/shSBDS-1; S-3, HeLa/shSBDS-3)
Discussion
Our current and previous studies [7, 8] showed that SBDS-deficiency in both primary hematopoietic cells and in shRNA-mediated SBDS-knockdown cells leads to increased apoptosis and hypersensitivity to Fas stimulation, suggesting that SBDS might be an important factor in regulating the Fas-mediated apoptosis pathway. To identify the molecular steps that lead to Fas hypersensitivity in SBDS-deficient cells we studied herein whether SBDS loss results in specific activation of the Fas pathway or in a general hypersensitivity to apoptosis stimuli. Although the TNF-α signaling pathway shares several factors with the Fas signaling pathway, the SBDS-knockdown cells did not show significant hypersensitivity to TNF-α, suggesting that the mechanism for the hypersensitivity is at the receptor level. Also, no hypersensitivity was observed to four different DNA damaging agents in both SBDS-knockdown cells and primary SDS hematopoietic progenitor cells which mainly activate the Bax apoptosis protein through p53. This agrees with our previous data showing no significant apoptosis through the Bax/Bcl-2/Bcl-XL pathway primary SDS cells and in SBDS-knockdown cells [8].
Recent studies on the SBDS orthologs suggested that SBDS is involved in RNA processing and ribosome biogenesis [12, 13]. If these postulated functions of SBDS account for the accelerated apoptosis in SBDS-deficient cells, one would expect that inhibition of either RNA transcription or protein synthesis would results in marked cell death. Notably, SBDS-knockdown cells were not hypersensitive to these agents, suggesting that these postulated functions might be independent of the SBDS role in regulating apoptosis through the Fas pathway in our cell model. In clonogenic assays, protein synthesis inhibition by cycloheximide did not results in differences between patients and controls. Treatment of SDS CD34+ cells with 0.5 μg/ml of α-amanitin resulted in slightly lower colony numbers compared to CD34+ from healthy subjects. However, the difference between patients and controls was not as marked as that observed with Fas stimulation, and significant overlap occurred between the two groups. Thus, whether primary marrow CD34+ cells are hypersensitive to low dose α-amanitin remains to be elucidated.
Although shRNA is now widely used as a powerful tool to knock down genes and study their functions, caution should be exerted not to suppress non-targeted genes [36, 37]. In our experiments, it is unlikely that the obtained results were due to nonspecific effects of the shRNA expression cassettes because the scrambled RNA control showed similar phenotype to the WT cells, and our two shRNAs were carefully designed against two different segments of SBDS gene and led to similar results. Further, our shRNA knockdown cell models manifest very similar apoptotic phenotype to primary SDS patient cells.
It is noteworthy that our SBDS-knockdown cells and the scrambled RNA-expressing control cells were established using a standard protocol by selection with geneticin, which is a protein synthesis inhibitor. However, as the vector used for transfection also carries a geneticin resistance gene, and as the HeLa/shSCR control cells had similar phenotype to the wild type cells, it is unlikely that the selection process had any effect on protein synthesis.
Since the SBDS-knockdown cells are specifically hypersensitive to Fas stimulation, the underlying mechanism of the hypersensitivity should reside in molecules related to the Fas pathway. It has been reported that the expression of Fas-related molecules such as Fas, FADD, caspase-8 and c-FLIP can affect the sensitivity to Fas stimulation [38–42]. Our data clearly demonstrate that Fas abnormally accumulates at the plasma membrane in SBDS-knockdown cells as well as in primary marrow cells. In contrast, FADD, procaspase-8 and procaspase-3 are not overexpressed. These later finding is in agreement with the observation that the SBDS-knockdown cells do not exhibit significant hypersensitivity to TNF-α, which uses the same proteins to executer apoptosis. We also analyzed the expression of the Fas signaling pathway inhibitors: XIAP, Erk and c-FLIP. There was no difference in the expression of XIAP, which inhibits caspase-3, -9 and possibly -8 [43, 44]. The levels of total Erk and phosphorylated Erk levels, which suppresses the Fas pathway by inhibition of caspase-8 [30, 45] were not reduced in the SBDS-deficient cells. Similarly, the levels of c-FLIPS and c-FLIPL, which compete with caspase-8 binding to FADD, [39] were not decreased in the SBDS-knockdown cells. From these experiments, we conclude that Fas hypersensitivity resides in Fas accumulation at the plasma membrane level. These latter two findings are in agreement with predominant expression of Fas transcript 1 in SBDS-deficient cells, which gives rise to the main Fas isoform containing a transmembrane domain and a death domain. Future studies with larger number of patients will enable accurate determination of the relative expression of this transcript to other transcripts, and whether the slight increase in Fas transcript 1 expression in SBDS-deficient cells is due to increased transcription, alternative splicing or enhanced stability. Another potential mechanism for increased accumulation of Fas at the cell membrane levels and hypersensitivity is increased trafficking. The expression of PI3K, Akt, Jak2, Stat1, and pStat1, which induce Fas trafficking in vascular smooth muscle cells [46], was not upregulated in SBDS-knockdown cells. Western blotting showed no differences in the protein expression of FAP1, which promotes Fas trafficking, and Dyn1/Dyn2, which inhibit trafficking, although enhanced activation of these proteins is still possible.
The results of our study suggest that loss of SBDS protein results in abnormal subcellular redistribution of Fas with specific localization to the plasma membrane, where it sensitizes the cells to stimulation by Fas ligand. Although SBDS has been postulated to function in ribosome biogenesis, many of the genes involved in ribosome biogenesis, including the Diamond–Blackfan anemia related gene, RPS19, are multifunctional, and many of them are involved in cell cycle and apoptosis. Therefore, it is possible that SBDS is another protein that plays a role in both ribosome biogenesis and apoptosis. Alternatively, the yet unknown function of SBDS in ribosome biogenesis might be related to proteins that also regulate apoptosis through the Fas pathway. It is possible that Fas-mediated apoptosis is at least in part responsible for the reduced cellularity in the bone marrow and exocrine pancreas of SDS patients. Inhibitors of Fas or its accumulation at the cell membrane might be used in preclinical models to study their ability to improve cell growth in SDS.
References
Shwachman H (1964) The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr 65:645–663. doi:10.1016/S0022-3476(64)80150-5
Dror Y (2005) Shwachman-Diamond syndrome. Pediatr Blood Cancer 45:892–901. doi:10.1002/pbc.20478
Donadieu J, Leblanc T, Bader Meunier B et al (2005) Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Haematologica 90:45–53
Majeed F, Jadko S, Freedman MH et al (2005) Mutation analysis of SBDS in pediatric acute myeloblastic leukemia. Pediatr Blood Cancer 45:920–924. doi:10.1002/pbc.20416
Smith OP, Hann IM, Chessells JM et al (1996) Haematological abnormalities in Shwachman-Diamond syndrome. Br J Haematol 94:279–284. doi:10.1046/j.1365-2141.1996.d01-1788.x
Boocock GR, Morrison JA, Popovic M et al (2003) Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet 33:97–101. doi:10.1038/ng1062
Dror Y, Freedman MH (2001) Shwachman-Diamond syndrome marrow cells show abnormally increased apoptosis mediated through the Fas pathway. Blood 97:3011–3016. doi:10.1182/blood.V97.10.3011
Rujkijyanont P, Watanabe KI, Ambekar C et al (2008) Sbds-deficient cells undergo accelerated apoptosis through the Fas pathway and not through the Bax.Bcl–2/Bcl-XL pathway. Haematologica 93:363–371. doi:10.3324/haematol.11579
Austin KM, Leary RJ, Shimamura A (2005) The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood 106:1253–1258. doi:10.1182/blood-2005-02-0807
Wu LF, Hughes TR, Davierwala AP et al (2002) Large-scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat Genet 31:255–265. doi:10.1038/ng906
Koonin EV, Wolf YI, Aravind L (2001) Prediction of the archaeal exosome and its connections with the proteasome and the translation and transcription machineries by a comparative-genomic approach. Genome Res 11:240–252. doi:10.1101/gr.162001
Savchenko A, Krogan N, Cort JR et al (2005) The Shwachman-Bodian-Diamond syndrome protein family is involved in RNA metabolism. J Biol Chem 280:19213–19220. doi:10.1074/jbc.M414421200
Shammas C, Menne TF, Hilcenko C et al (2005) Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond syndrome. J Biol Chem 280:19221–19229. doi:10.1074/jbc.M414656200
Dror Y, Durie P, Ginzberg H et al (2002) Clonal evolution in marrows of patients with Shwachman-Diamond syndrome: a prospective 5-year follow-up study. Exp Hematol 30:659–669. doi:10.1016/S0301-472X(02)00815-9
Stepanovic V, Wessels D, Goldman FD et al (2004) The chemotaxis defect of Shwachman-Diamond Syndrome leukocytes. Cell Motil Cytoskeleton 57:158–174. doi:10.1002/cm.10164
Wessels D, Srikantha T, Yi S et al (2006) The Shwachman-Bodian-Diamond syndrome gene encodes an RNA-binding protein that localizes to the pseudopod of Dictyostelium amoebae during chemotaxis. J Cell Sci 119:370–379. doi:10.1242/jcs.02753
Dror Y, Freedman MH (1999) Shwachman-Diamond syndrome: An inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment. Blood 94:3048–3054
Rathbun RK, Christianson TA, Faulkner GR et al (2000) Interferon-gamma-induced apoptotic responses of Fanconi anemia group C hematopoietic progenitor cells involve caspase 8-dependent activation of caspase 3 family members. Blood 6:4204–4211
Cumming RC, Lightfoot J, Beard K et al (2001) Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat Med 7:814–880. doi:10.1038/89937
Perdahl EB, Naprstek BL, Wallace WC et al (1994) Erythroid failure in Diamond-Blackfan anemia is characterized by apoptosis. Blood 83:645–650
Flygare J, Kiefer T, Miyake K et al (2005) Deficiency of ribosomal protein S19 in CD34+ cells generated by siRNA blocks erythroid development and mimics defects seen in Diamond-Blackfan anemia. Blood 105:4627–4634. doi:10.1182/blood-2004-08-3115
Carlsson G, Aprikyan AA, Tehranchi R et al (2004) Kostmann syndrome: severe congenital neutropenia associated with defective expression of Bcl-2, constitutive mitochondrial release of cytochrome c, and excessive apoptosis of myeloid progenitor cells. Blood 103:3355–3361. doi:10.1182/blood-2003-04-1011
Itoh N, Yonehara S, Ishii A et al (1991) The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66:233–243. doi:10.1016/0092-8674(91)90614-5
Fas NS (1999) ligand-induced apoptosis. Annu Rev Genet 33:29–55. doi:10.1146/annurev.genet.33.1.29
Lepelley P, Grardel N, Erny O et al (1998) Fas/APO-1 (CD95) expression in myelodysplastic syndromes. Leuk Lymphoma 30:307–312
Piazzolla D, Meissl K, Kucerova L et al (2005) Raf-1 sets the threshold of Fas sensitivity by modulating Rok-alpha signaling. J Cell Biol 171:1013–1022. doi:10.1083/jcb.200504137
Dror Y, Freedman MH (2002) Shwachman-diamond syndrome. Br J Haematol 118:701–713. doi:10.1046/j.1365-2141.2002.03585.x
Wojcik C, Fabunmi R, DeMartino GN (2005) Modulation of gene expression by RNAi. Methods Mol Med 108:381–393
Holmstrom TH, Tran SE, Johnson VL et al (1999) Inhibition of mitogen-activated kinase signaling sensitizes HeLa cells to Fas receptor-mediated apoptosis. Mol Cell Biol 19:5991–6002
Tran SE, Holmstrom TH, Ahonen M et al (2001) MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J Biol Chem 276:16484–16490. doi:10.1074/jbc.M010384200
Rujkijyanont P, Beyene J, Wei K et al (2007) Leukaemia-related gene expression in bone marrow cells from patients with the preleukaemic disorder Shwachman-Diamond syndrome. Br J Haematol 137:537–544. doi:10.1111/j.1365-2141.2007.06608.x
Tsai PH, Sahdev I, Herry A et al (1990) Fatal cyclophosphamide-induced congestive heart failure in a 10-year-old boy with Shwachman-Diamond syndrome and severe bone marrow failure treated with allogeneic bone marrow transplantation. Am J Pediatr Hematol Oncol 12:472–476. doi:10.1097/00043426-199024000-00012
Okcu F, Roberts WM, Chan KW (1998) Bone marrow transplantation in Shwachman-Diamond syndrome: report of two cases and review of the literature. Bone Marrow Transplant 121:849–851. doi:10.1038/sj.bmt.1701170
Cesaro S, Oneto R, Messina C et al (2005) EBMT Severe Aplastic Anaemia and Paediatric Diseases Working Party. Haematopoietic stem cell transplantation for Shwachman-Diamond disease: a study from the European Group for blood and marrow transplantation. Br J Haematol 131:231–236. doi:10.1111/j.1365-2141.2005.05758.x
Li JH, Kluger MS, Madge LA et al (2002) Interferon-gamma augments CD95 (APO-1/Fas) and pro-caspase-8 expression and sensitizes human vascular endothelial cells to CD95-mediated apoptosis. Am J Pathol 161:1485–1495
Jackson AL, Bartz SR, Schelter J et al (2003) Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 21:635–637. doi:10.1038/nbt831
Snove O Jr, Holen T (2004) Many commonly used siRNAs risk off-target activity. Biochem Biophys Res Commun 319:256–263. doi:10.1016/j.bbrc.2004.04.175
Hougardy BM, van der Zee AG, van den Heuvel FA et al (2005) Sensitivity to Fas-mediated apoptosis in high-risk HPV-positive human cervical cancer cells: relationship with Fas, caspase–8, and Bid. Gynecol Oncol 97:353–364. doi:10.1016/j.ygyno.2005.01.036
Scaffidi C, Schmitz I, Krammer PH et al (1999) The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 274:1541–1548. doi:10.1074/jbc.274.3.1541
Schneider EM, Menzl I, Weber O et al (2003) Differential calcium response in HeLa and HeLa-Fas cells by cytotoxic T lymphocytes. Biochem Biophys Res Commun 301:159–166. doi:10.1016/S0006-291X(02)02968-6
Snow AL, Chen LJ, Nepomuceno RR et al (2001) Resistance to Fas-mediated apoptosis in EBV-infected B cell lymphomas is due to defects in the proximal Fas signaling pathway. J Immunol 167:5404–5411
Willems F, Amraoui Z, Vanderheyde N et al (2000) Expression of c-FLIP(L) and resistance to CD95-mediated apoptosis of monocyte-derived dendritic cells: inhibition by bisindolylmaleimide. Blood 95:3478–3482
Takahashi R, Deveraux Q, Tamm I et al (1998) A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem 273:7787–7790. doi:10.1074/jbc.273.14.7787
Sun C, Cai M, Meadows RP et al (2000) NMR structure and mutagenesis of the third Bir domain of the inhibitor of apoptosis protein XIAP. J Biol Chem 275:33777–33781. doi:10.1074/jbc.M006226200
Irmler M, Thome M, Hahne M et al (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388:190–195. doi:10.1038/40657
Rosner D, Stoneman V, Littlewood T et al (2006) Interferon-γ induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K- and Akt-depndent mechanism. Am J Pathol 168:2054–2063. doi:10.2353/ajpath.2006.050473
Acknowledgment
The work was supported by grants from the Canadian Institute of Health Research MOP57720, Shwachman–Diamond Support Canada, and Shwachman–Diamond Syndrome Projects LTD. KIW is a recipient of the Hospital for Sick Children Research Training Award.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ken-ichiro Watanabe and Chhaya Ambekar contributed equally to the article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Watanabe, Ki., Ambekar, C., Wang, H. et al. SBDS-deficiency results in specific hypersensitivity to Fas stimulation and accumulation of Fas at the plasma membrane. Apoptosis 14, 77–89 (2009). https://doi.org/10.1007/s10495-008-0275-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0275-9