
Abstract
There is increasing evidence that the active contribution of hepatocytes to liver disease is strongly dependent on local cytokine environment. It has been shown in vitro that TNFα can enhance hepatocyte FasLigand (FasL)-mediated cytotoxicity. Here, we demonstrate that TNFα-induced apoptosis was associated with Fas and FasL upregulation and that a FasL-neutralizing antibody prevented TNFα-induced apoptosis. We further examined in vivo the relevance of the Fas/FasL pathway to hepatocellular apoptosis in a TNFα-driven model of acute liver failure. Livers of galactosamine/lipopolysaccharide (Gal/LPS)-exposed Fas wild-type mice highly expressed both Fas and FasL and revealed marked hepatocellular apoptosis that was almost completely blocked by soluble TNFα-receptor; this was also almost absent in Gal/LPS-exposed Fas lymphoproliferation mutant mice. Our data provide evidence for a direct link between TNFα and Fas/FasL in mediating hepatocyte apoptosis. Fratricidal death by Fas–FasL interactions of neighbouring hepatocytes may actively contribute to acute liver failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute liver failure (ALF) is a rare clinical syndrome associated with high mortality. Hepatic failure leads to a well-recognized pattern of clinical signs and symptoms, sometimes with rapid deterioration and progression to multi-organ failure [1]. Hepatic cell death is the leading cause of fatality in patients with ALF. The development of hepatic necrosis and apoptosis gives rise to severe hyperammonemia, hepatic encephalopathy, and life-threatening cerebral edema. In contrast with the well-documented morphological characterization of dying hepatocytes, the molecular pathways leading to the death of hepatocytes are not completely understood. A large amount of recent research indicates that a complex system, comprising death factors and death receptors, mediates cell death in liver disease. Our understanding of which cell death receptors are involved and how they function will become increasingly important for developing rational therapeutic strategies. In line with this, anti-apoptotic interventions have been stated to be among the most promising pharmacological options in the near future to ameliorate clinical liver disease [2].
Death receptors that have been assessed in purified liver cell preparations include Fas, Toll-like receptors (TLRs), and tumor necrosis factor receptors (TNFR)-1 and -2, as well as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptors 1, 2, 3, and 4 [3]. Accordingly, the endogenous production of FasLigand (FasL) and TNFα has been implicated in mediating hepatic cell death in experimental models of hepatitis. Blockade of death receptor pathways has been shown to ameliorate liver disease to various degrees [4–8]. Within this context, however, the relative contribution of these pathways to hepatic injury is controversial and seems to depend significantly on the experimental models used. While in concanavalin A-induced hepatitis the contribution of TNFα to hepatic injury appears to predominate [5], other groups have failed to confirm the role of TNFα in this model and instead suggest a FasL-dependent pathway [9, 10]. In contrast with these findings, the galactosamine/lipopolysaccharide (Gal/LPS)-induced model of liver injury is reported to be mediated by neither Fas antigen [8] nor FasL [6]; however, it seems to unequivocally comprise the secretion of TNFα and its binding to TNFR-1, thereby mediating hepatic injury and lethality [11].
Most recently, hepatocytes have been recognized as cytotoxic effector cells under conditions of inflammation. Within this context, it was reported that hepatocytes exposed to TNFα exert cytotoxic activity via a FasL-mediated pathway and eliminate Fas-sensitive target cells [12]. Based on these findings, it is reasonable to hypothesize that under conditions of hepatic inflammation, such as Gal/LPS-induced ALF with TNFα release, hepatocyte Fas/FasL-dependent killing might contribute to liver injury in either a fratricidal or a suicidal manner [12, 13]. To address the relevance of upregulation of FasL and its cognate receptor in hepatocyte cytotoxic activity, we studied Fas/FasL expression and hepatocellular apoptosis in Gal/LPS-exposed Fas wild-type (Fas wt) and Fas lpr (lymphoproliferation) mutant (Fas lpr/lpr) mice and neutralized TNFα by application of a soluble TNFα-receptor (sTNFα-R).
Material and methods
Cytochemistry and flow cytometry of HepG2 cells
The human hepatoma cell line HepG2 was used for all in vitro experiments and was cultured as reported [12]. Cells were seeded in four-well Chamber Slides (LAB-TEKT; Nunc, Wiesbaden, Germany) for cytochemical analysis and in six-well plates for flow cytometric analysis. After reaching confluence, cells were incubated with 500 U/mL human TNFα (hTNFα; Sigma, Taufkirchen, Germany) or vehicle solution (n = 4 independent experiments each) for 6 h to induce apoptosis, in accordance with previously published work [12]. In an additional series of experiments, we simultaneously added 200 ng/mL mAb NOK-1 (BD Pharmingen, Heidelberg, Germany) to neutralize FasL.
For direct visualization of TNFα-induced apoptotic cell death cells were incubated with bisbenzimide (Hoechst 33342; Sigma) for an additional 15 min at room temperature (RT). The labeled cells were analyzed with a fluorescence microscope (Axioskop 40; Zeiss, Jena, Germany).
Flow cytometric analysis of cell apoptosis was performed by means of 7-AAD staining as described by Philpott et al. [14]. After cell detachment by trypsinization and washing, cells were fixed in 70% methanol at −20°C for 20 min, followed by incubation with 25 μg/ml RNase A (Roth, Karlsruhe, Germany) for 15 min at 37°C and staining with 7-AAD (200 μg/ml; Molecular Probes, Eugene, OR, USA) for 30 min at RT. Apoptotic cells were assessed by quantitative analysis of the sub G0/G1 peak using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA).
For evaluation of concomitant Fas and FasL expression, fixed cells were incubated with rabbit polyclonal anti-Fas (1:100, M-20; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit polyclonal anti-FasL (1:100, C-20; Santa Cruz Biotechnology), or with an isotype-specific immunoglobulin (1:200; Santa Cruz Biotechnology) as a negative control antibody for 30 min at 37°C. After two washing steps, goat anti-rabbit–FITC antibody (1:200; Santa Cruz Biotechnology) was applied for 30 min at 37°C. The labeled cells were analyzed by means of FACScan flow cytometry.
Model of ALF and experimental groups
Male Fas wild-type mice (C57BL/6J; Fas wt) (Charles River Laboratories, Sulzfeld, Germany) and Fas lpr mutants (B6-lpr/lpr(Fas); Fas lpr/lpr) (Jackson Laboratory, Maine, USA) were used at 8–10 week of age with a body weight of approximately 20 g. Animals were provided with water and standard laboratory chow ad libitum. The experimental protocol was approved by the local committee and all animals received humane care according to the German legislation on protection of animals and the Guide for the Care and Use of Laboratory Animals (NIH publication 86-23 revised 1985).
For induction of ALF, mice were injected with Gal (720 mg/kg body weight intraperitoneally [bw ip]; Sigma) and LPS (10 μg/kg bw ip, serotype 0128:B12; Sigma) and were studied 6 h thereafter. Concentrations of Gal and LPS were used in accordance with work published previously by our [15, 16] and other groups [8, 17]. To address the contribution of TNFα in Gal/LPS-induced ALF, animals were pretreated with sTNFα-R (recombinant human TNFR Fc fusion protein, etanercept, 100 μg/kg bw ip, Enbrel; Wyeth Pharma GmbH, Münster, Germany) or equivalent volumes of vehicle solution (0.9% saline 1 mL/kg bw ip) 1 h prior to ALF induction. Although it is of human origin, etanercept has been characterized as a TNFα-blocking drug that also prevents rheumatoid arthritis in mice [18].
Intravital fluorescence microscopy
For in vivo analysis of hepatocellular apoptosis, intrahepatic leukocyte accumulation, and sinusoidal perfusion failure, fluorescence microscopy was performed 6 h after Gal/LPS exposure in ketamine/xylazine-anesthetized animals (75/25 mg/kg bw ip) in accordance with work previously published by our group [15, 16, 19].
Sampling and assays
After in vivo microscopy, animals were exsanguinated by puncture of the vena cava inferior for immediate separation of EDTA plasma. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activities were measured spectrophotometrically as indicators for hepatocellular disintegration and necrosis. Liver tissue was sampled for subsequent RT-PCR, Western blot analysis, histology, and immunohistochemistry.
RT-PCR analysis of liver tissue
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer’s instructions. cDNA synthesis was performed using Superscript II RT (Invitrogen, Karlsruhe, Germany) in accordance with the manufacturer’s instructions. PCR for murine Fas, FasL, and GAPDH was performed in a 20 μL reaction volume containing 1× reaction puffer, 20 μM dNTPs, and 5,000 U/mL Taq polymerase. Samples were incubated with respective primers (Table 1) for either 30 (FasL and GAPDH) or 35 (Fas) cycles (denaturation for 30 s at 94°C; annealing for 30 s at 58°C (Fas), 56°C (FasL), or 54°C (GAPDH); and elongation for 30 s at 72°C) using a thermocycler (Eppendorf Mastercycler Gradient, New York, NY, USA). PCR products were analyzed on a 1% agarose gel (peqGOLD Universal Agarose; peQLab Biotechnologie, Erlangen, Germany) containing 0.5 μg/ml ethidium bromide. Signals were densitometrically assessed (Quantity One, ChemiDoc XRS System; Bio-Rad Laboratories, Munich, Germany) and normalized to the GAPDH signals.
Western blot analysis of liver tissue
For Western blot analysis of protein levels of cleaved caspase-3, Fas, and FasL, liver tissue was homogenized in lysis buffer (10 mM Tris pH 7.5, 10 mM NaCl, 0.1 mM EDTA, 0.5% Triton-X 100, 0.02% NaN3, 0.2 mM PMSF, and protease inhibitor cocktail), incubated for 30 min on ice and centrifuged for 15 min at 10,000g. Protein content were assayed by the BCA method (Pierce Biotechnology, Rockford, IL, USA) with bovine serum albumin (BSA) as the standard. Per lane, either 40 μg of protein (cleaved caspase-3 and TLR-4) or 20 μg of protein (Fas and FasL) was separated on a 12% SDS gel and transferred to a polyvinyldifluoride membrane (Immobilon-P; Millipore, Eschborn, Germany). After blocking with 2% BSA (Sigma), membranes were incubated for 2 h at RT with a rabbit polyclonal anti-cleaved caspase 3 (1:1,000, Asp 175; Stressgen Biotech, San Diego, CA, USA), rabbit polyclonal anti-FasL (1:500, C-20; Santa Cruz Biotechnology), rabbit polyclonal anti-Fas (1:500, M-20; Santa Cruz Biotechnology), and goat polyclonal anti-TLR-4 (1:1000, M-16; Santa Cruz Biotechnology) followed by a secondary peroxidase-linked goat anti-rabbit antibody (cleaved caspase-3 and FasL: 1:2,000 and Fas: 1:5,000; Cell Signaling Technology, Boston, MA, USA) and donkey anti-goat antibody (TLR-4: 1:7,500; Santa Cruz Biotechnology). Protein expression was visualized by means of luminol-enhanced chemiluminescence (ECL plus; Amersham Pharmacia Biotech, Freiburg, Germany) and digitalized with the ChemiDoc XRS System (Bio-Rad Laboratories GmbH). Signals were densitometrically assessed (Quantity One; Bio-Rad Laboratories) and normalized to β-actin signals (mouse monoclonal anti-β-actin antibody: 1:20,000; Sigma).
Histology and immunohistochemistry of liver tissue
For hematoxylin and eosin (H&E) staining for the immunohistochemical study of cleaved caspase-3 and for analysis of TUNEL-positive hepatocytes, we used protocols in accordance with work previously published by our group [15, 16]. For immunhistochemical staining of Fas and FasL expression serial sections of liver tissue were incubated with primary antibodies overnight at 4°C (polyclonal anti-Fas (1:50, M-20) and polyclonal anti-FasL (1:50, C-20); both Santa Cruz Biotechnology). Universal LSAB® kits (System-HRP and System-AP; DakoCytomation, Dako, Hamburg, Germany) were used according to the manufacturer’s instructions for the development of Fas with DAB chromogen and of FasL with fuchsin chromogen. The sections were counterstained with hemalaun and analyzed with a light microscope (Olympus BX51, Hamburg, Germany).
Statistical analysis
All data are expressed as mean ± SEM. Statistical differences among groups were determined using an unpaired Student’s t-test. Data were considered significant when P < 0.05. Statistical analysis was performed using the Sigma Stat and Sigma Plot software package (Jandel Scientific, San Rafael, CA, USA).
Results
Cytochemistry and flow cytometry of HepG2 cells
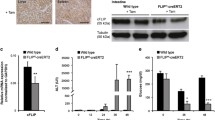
Under basal conditions, flow cytometry of HepG2 cultures demonstrated that 4% of the cells underwent apoptosis (Fig. 1a, b) with a negligible fraction concomitantly expressing Fas (~1%) or FasL (~5%). In contrast, TNFα exposure of HepG2 cells resulted in a huge rise in apoptotic cells (57 ± 11%), of which 11 ± 2% expressed Fas, but 44 ± 8% expressed FasL (Fig. 1c). Noteworthy, adding the FasL-neutralizing antibody NOK-1 reduced TNFα-induced apoptosis to 9 ± 1% (Fig. 1b), of these apoptotic cells, only 20 ± 3% now expressed FasL (Fig. 1c). These findings underscore the involvement of the Fas/FasL pathway in the TNFα-dependent apoptosis of HepG2 cells.

Representative UV fluorescence images (a, upper) of bisbenzimide-stained HepG2 cells as well as images of the corresponding fields in trans-illumination technique (a, lower) upon hTNFα exposure (original magnification 500×). Note the typical signs of apoptotic HepG2 cells, i.e., nuclear fragmentation and condensation. Flow cytometric analysis of HepG2 cells (b) upon induction of apoptosis by human TNFα exposure and additional treatment with the FasL-neutralizing mAb NOK-1. Percentage of apoptotic HepG2 cells expressing either Fas or FasL (c). Values are given as mean ± SEM; n = 4 independent experiments; unpaired Student’s t-test, including Bonferroni correction to compensate for multiple comparisons; *P < 0.05 vs. unstimulated, # P < 0.05 vs. hTNFα
Expression of Fas and FasL in Gal/LPS-exposed livers
RNA analysis of tissue from Gal/LPS-exposed livers showed a 1.4-fold increase in Fas expression and a 2-fold increase in FasL expression in Fas wt mice when compared with controls, i.e., animals without Gal/LPS challenge (Fig. 2a, b). TNFα blockade elicits a reduction in Fas and FasL mRNA expression of ~30% in Fas wt mice (Fig. 2a, b). In Fas lpr/lpr mice Fas mRNA was not detectable (Fig. 2a), but FasL mRNA expression was also upregulated after Gal/LPS exposition (Fig. 2b).

Representative RT-PCR gels and densitometric analysis of Fas (a) and FasL (b) mRNA levels in livers of Fas wt (n = 8) and Fas lpr/lpr (n = 8) mice. Signals were corrected using GAPDH signal. Animals were injected with Gal and LPS for induction of ALF (−6 h) and pretreated (−1 h) with either soluble recombinant TNFα-R (sTNFα-R; n = 4) or equivalent volumes of vehicle solution (vehicle; n = 4). Control represents animals without induction of ALF receiving only isotonic saline. Values are given as mean ± SEM; unpaired Student’s t-test; # P < 0.05 vs. vehicle; n.d. = not detectable
Protein analysis of Gal/LPS-exposed liver tissue showed a 3.6-fold increase in Fas expression and a 1.6-fold increase in FasL expression in Fas wt mice compared with controls, and revealed a marked reduction in both Fas (−39%) and FasL (−50%) upon TNFα blockade in Fas wt mice (Fig. 3a, b), indicating the dependence of the Fas/FasL pathway on TNFα during ALF. Fas lpr/lpr mice exhibited significantly lower levels of Fas and FasL compared with those found in Fas wt mice, and showed a significant reduction in Fas, but not in FasL, upon TNFα neutralization (Fig. 3a, b).

Representative Western blots and densitometric analysis of Fas (a) and FasL (b) protein levels in livers of Fas wt (n = 8) and Fas lpr/lpr animals (n = 8). Signals were corrected using β-actin signal. Animals were injected with Gal and LPS for induction of ALF (−6 h) and pretreated (−1 h) with either soluble recombinant TNFα-R (sTNFα-R; n = 4) or equivalent volumes of vehicle solution (vehicle; n = 4). Control represents animals without induction of ALF receiving only isotonic saline. Values are given as mean ± SEM; unpaired Student’s t-test; *P < 0.05 vs. Fas wt; # P < 0.05 vs. vehicle
Additionally, immunohistochemical staining of Fas and FasL of serial liver tissue sections revealed a marked hepatocellular expression of the receptor and its ligand in the Gal/LPS-exposed Fas wt mice (Fig. 4, left panels), while only a weak staining of both Fas and FasL was detectable in the respective Fas lpr/lpr mice (Fig. 4, right panels). Noteworthy, fratricidal cell death by Fas–FasL interaction is underscored by the fact that Fas positive hepatocytes were found in the direct neighbourhood of hepatocytes which were FasL positive.

Representative light microscopic images (upper panels, original magnification 200×; middle, 400×; lower, 1,000×) of immunohistochemical staining of Fas (DAB brown staining) and FasL (fuchsin red staining) in serial liver tissue sections of Fas wt (left panels) and Fas lpr/lpr mice (right panels). Note the marked hepatocellular expression of either Fas (asterisk) or FasL (arrow head) of neighbouring hepatocytes in Gal/LPS-exposed Fas wt mice (left panel, 1,000×) and the nuclear chromatin fragmentation of the FasL positive hepatocyte as a sign of apoptosis indicating fratricidal cell death by Fas–FasL interaction. In contrast, there is only weak staining of both Fas and FasL in the respective Fas lpr/lpr mice (right panel)
Furthermore, Gal/LPS exposure with and without sTNFα-R revealed a 2-fold rise in TLR-4 protein expression in the liver tissue of Fas wt mice compared with the control (Fig. 5). In contrast, Gal/LPS-exposed Fas lpr/lpr mice exhibited only ~10% of the TLR-4 expression seen in Fas wt mice (Fig. 5).

Representative Western blots and densitometric analysis of TLR-4 protein levels in livers of Fas wt (n = 8) and Fas lpr/lpr animals (n = 8). Signals were corrected using β-actin signal. Animals were injected with Gal and LPS for induction of ALF (−6 h) and pretreated (−1 h) with either soluble recombinant TNFα-R (sTNFα-R; n = 4) or equivalent volumes of vehicle solution (vehicle; n = 4). Control represents animals without induction of ALF receiving only isotonic saline. Values are given as mean ± SEM; unpaired Student’s t-test; *P < 0.05 vs. Fas wt
Fas lpr/lpr mice showed a decrease in ALF-associated apoptotic cell death
An analysis of apoptotic cell death after Gal/LPS exposure is provided in Figs. 6 and 7, including in vivo microscopy (Fig. 6) and TUNEL histochemistry (Fig. 7) of hepatocellular apoptosis.

Representative fluorescent microscopic images (upper panel, original magnification 200×) and quantitative analysis (lower panel) of apoptotic hepatocytes in Fas wt (n = 14) and Fas lpr/lpr animals (n = 14). Animals were injected with Gal and LPS for induction of ALF (−6 h) and pretreated (−1 h) with either soluble recombinant TNFα-R (sTNFα-R; n = 7) or equivalent volumes of vehicle solution (vehicle; n = 7). Hepatocellular apoptosis was assessed by in vivo fluorescence microscopy and bisbenzimide staining. Compare the high numbers of individual hepatocytes with condensation, but also fragmentation and/or margination of nuclear chromatin, characteristic signs of apoptotic cell death in Fas wt mice (upper left image), with the few apoptotic hepatocytes found in Fas wt mice upon TNFα blockade (upper right image), and in Fas lpr/lpr mice (lower left and right images). Values are given as mean ± SEM; unpaired Student’s t-test; *P < 0.05 vs. Fas wt; # P < 0.05 vs. vehicle; § P < 0.05 vs. Fas wt/sTNFα-R

Representative light microscopic images (upper panel, original magnification 200×) and quantitative analysis (lower panel) of TUNEL histochemistry for apoptotic hepatocytes in Fas wt (n = 14) and Fas lpr/lpr animals (n = 14). Animals were injected with Gal and LPS for induction of ALF (−6 h) and pretreated (−1 h) with either soluble recombinant TNFα-R (sTNFα-R; n = 7) or equivalent volumes of vehicle solution (vehicle; n = 7). Note the high numbers of TUNEL-positive hepatocytes in Fas wt mice (upper left image) and the marked reduction in cell apoptosis in Fas wt mice upon TNFα blockade (upper right image), and in Fas lpr/lpr mice (lower left and right images). Values are given as mean ± SEM; unpaired Student’s t-test; *P < 0.05 vs. Fas wt; # P < 0.05 vs. vehicle; § P < 0.05 vs. Fas wt/sTNFα-R
Fas wt mice exposed to Gal/LPS were characterized by a high number of apoptotic hepatocytes, while Fas lpr/lpr mice were markedly protected against ALF-associated apoptotic liver injury (Fig. 6). Confirming this, TUNEL histochemistry of liver tissue from Fas lpr/lpr mice with ALF showed a more than 60% reduction in TUNEL-positive cells (Fig. 7). Gal/LPS-induced liver injury was found to be responsive to TNFα blockade, because pretreatment with sTNFα-R resulted in nearly complete inhibition of apoptosis in Fas wt mice with a negligible low number of either bisbenzimide-stained hepatocytes, exhibiting nuclear chromatin condensation and fragmentation (Fig. 6), or TUNEL-positive hepatocytes (Fig. 7). Of utmost interest, the extent of hepatocellular apoptosis in Gal/LPS-challenged and vehicle-treated Fas lpr/lpr mice was significantly higher than that in Gal/LPS-challenged and TNFα-blocked Fas wt animals. This indicates that both the TNFα and the Fas/FasL pathway contribute to apoptotic injury in ALF. In further support of this view, TNFα neutralization in Fas lpr/lpr mice reduced the number of apoptotic hepatocytes to values similar to those found in TNFα-blocked Fas wt mice (Figs. 6, 7).
In support of these data, immunohistochemical and protein analysis of cleaved caspase-3 (Fig. 8) revealed less activation of the effector caspase in Fas lpr/lpr vs. Fas wt mice after Gal/LPS challenge and treatment with the vehicle (control). Additionally, Western immunoblots for cleaved caspase-3 confirmed the dependency of Gal/LPS-induced ALF on TNFα, as shown by the marked decrease in cleaved caspase-3 protein upon pretreatment with sTNFα-R in Fas lpr/lpr, but most particularly in Fas wt mice (Fig. 8). Furthermore, the blockade of TNFα in Fas wt mice was more effective with respect to preventing apoptosis than was the Fas lpr mutation, as indicated by the significantly higher cleavage of caspase-3 protein in these animals after Gal/LPS exposure or treatment with the vehicle (Fig. 8).

Representative light microscopic images (a, upper panel, original magnification 200×) and Western blot, as well as densitometric analysis (b), of cleaved caspase-3 protein levels in livers of Fas wt (n = 8) and Fas lpr/lpr animals (n = 8). Animals were injected with Gal and LPS for induction of ALF (−6 h) and pretreated with either soluble recombinant TNFα-R (sTNFα-R; n = 4) or equivalent volumes of vehicle solution (vehicle; n = 4). Values are given as mean ± SEM; unpaired Student’s t-test; *P < 0.05 vs. Fas wt; # P < 0.05 vs. vehicle; § P < 0.05 vs. Fas wt/sTNFα-R
Fas lpr mutation and TNFα blockade ameliorated hepatic microcirculatory dysfunction and liver tissue disintegration
Hepatic microcirculation analysis by fluorescence microscopy is given in Fig. 9. In the Gal/LPS-treated Fas wt mice, in vivo microscopy of the livers revealed characteristic features of acute injury, including increased sinusoidal leukocyte accumulation and pronounced perfusion failure of up to 59% nonperfused sinusoids (Fig. 9a, b). Application of sTNFα-R in Fas wt mice and Fas lpr mutation per se was associated with a reduction in inflammatory leukocyte stasis and microvascular perfusion failure (Fig. 9a, b).

Representative fluorescent microscopic images (original magnification 200×) and quantitative analysis of hepatic sinusoidal leukocyte stasis (a) and sinusoidal perfusion failure (b) in Fas wt (n = 14) and Fas lpr/lpr animals (n = 14). Animals were injected with Gal and LPS for induction of ALF (−6 h) and pretreated (−1 h) with either soluble recombinant TNFα-R (sTNFα-R; n = 7) or equivalent volumes of vehicle solution (vehicle; n = 7). Values are given as mean ± SEM; unpaired Student’s t-test; *P < 0.05 vs. Fas wt; # P < 0.05 vs. vehicle
H&E staining of liver tissue specimens from Gal/LPS-exposed Fas wt mice exhibited disruption of the general architecture and microvascular disintegration, as well as tissue apoptosis and necrosis (Fig. 10a). In line with the histopathology results, liver injury was reflected by high transaminase activities in plasma (Fig. 10b, c). Liver sections obtained from Fas wt mice with TNFα blockade and Fas lpr/lpr mice per se were different in appearance, often exhibiting grossly retained general architecture and lacking evidence of major morphological injury (Fig. 10a). Accordingly, transaminase levels were significantly reduced in these animals (Fig. 10b, c), although there were still significantly higher AST and ALT values in vehicle- and sTNFα-R-treated Fas lpr/lpr animals when compared with transaminase levels in the TNFα-blocked Fas wt mice (Fig. 10).

Representative H&E-stained images (a, original magnification 400×) and plasma AST (b) and ALT activities (c) in Fas wt (n = 14) and Fas lpr/lpr animals (n = 14). Animals were injected with Gal and LPS for induction of ALF (−6 h) and pretreated (−1 h) with either soluble recombinant TNFα-R (sTNFα-R; n = 7) or equivalent volumes of vehicle solution (vehicle; n = 7). a Note the deterioration of hepatic morphology, including tissue apoptosis (arrow-heads) and necrosis (arrows) in Gal/LPS-exposed Fas wt mice (left upper image), while tissue integrity was almost entirely maintained in liver specimens from animals in the other groups. Values are given as mean ± SEM; unpaired Student’s t-test; *P < 0.05 vs. Fas wt; # P < 0.05 vs. vehicle; § P < 0.05 vs. Fas wt/sTNFα-R
Discussion
In this study, we showed that the upregulation of Fas and FasL in Gal-sensitized mice upon exposure to LPS is dependent upon TNFα; this was shown by application of sTNFα-R, which neutralized TNFα, partially preventing inflammatory Fas and FasL expression. We further demonstrated the relative contribution of TNFα and the Fas/FasL pathway to hepatic injury and observed that both Fas lpr mutation and blockade of endogenous TNFα limited hepatic injury, although we found that neutralization of TNFα was more potent in preventing Gal/LPS-induced hepatocellular apoptosis. This led us to conclude that hepatocyte apoptosis is indeed driven mainly by TNFα; however, it necessarily includes Fas/FasL cytotoxicity in the present murine model of ALF. In addition, TNFα-induced apoptosis of HepG2 cells was substantially reduced by a neutralizing anti-FasL antibody. Thus, we propose that TNFα-dependent hepatocyte apoptosis involves activation of Fas by upregulation of FasL, and that Fas/FasL-mediated cytotoxicity of hepatocytes is of considerable relevance in the present TNFα-driven model of liver injury.
Methodological considerations
As reported by our group [15, 16] and other groups [8, 17], we used the administration of LPS to Gal-sensitized mice as an experimental model for endotoxic or septic shock [8, 11], although other models for liver injury-like application of TNFα to Gal-sensitized mice would also have been appropriate [17]. In contrast to previous predominantly T-cell-mediated models of hepatitis, Gal/LPS-induced liver injury is presumed to be principally a macrophage/monocyte-mediated model [20], and presents, in our hands, with high apoptotic cell death, deterioration of microcirculation, and transaminase release. We focused in the current work on mediation of hepatocellular apoptosis, which was confirmed by several independent methods, such as in vivo fluorescence microscopy and TUNEL histochemistry, as well as protein analysis and immunohistochemistry of caspase-3 cleavage.
While other groups have used mice with a spontaneous mutation in FasL or applied soluble Fas fusion protein [6], we used mice with the lpr mutation in the Fas gene to address the contribution of the Fas/FasL pathway in Gal/LPS-induced liver injury. In these mice, insertion of an early transposable element (ETn) in intron 2 of Fas antigen results in a loss-of-function mutation [21] with the consequence of a small but distinct rearrangement of the Fas antigen gene [22]. Although we were not able to detect Fas mRNA transcription, it has been reported that a small amount of transcript can be read through the ETn region and spliced correctly [23]. This might explain the detection of Fas protein in the Fas lpr/lpr mice of the present study, similar to what has been described by others [24].
Although we were aware of the possibility that liver tissue could be contaminated by infiltrating cells bearing Fas and FasL, we analyzed Fas and FasL by RT-PCR and Western blot analysis in samples of whole-liver tissue. Referring to this, real-time RT-PCR quantification of Fas and FasL in whole-liver could exclude contamination with infiltrating cells based on the absence of CD3 cDNA signals [12].
Mediation of hepatocyte apoptosis in Gal/LPS-exposed livers
Apoptosis of hepatocytes is known to be a major cause of hepatocellular injury in a variety of liver diseases. Neutralization of TNFα by either monoclonal antibodies, inhibitors of TNFα processing, or anti-TNFα pharmacologic agents suppresses mortality in liver injury [25]. Similarly, it has been reported that TNFα plays a crucial role in the lethal activity of Gal/LPS [26] and that it is a terminal mediator for it [27]. The evidence that LPS-induced apoptosis in Gal-sensitized mice is mediated by TNFα release [8, 27] is further substantiated by the fact that anti-TNFα antibody [8] and sTNFα-R (present study) almost completely prevented LPS-induced hepatocyte apoptosis in Gal-exposed mice. Moreover, apoptosis of hepatocytes can be reproduced by injecting recombinant TNFα [8, 17].
Furthermore, it is well known that LPS induces TNFα release by Kupffer cells in the presence of functioning TLR-4 [28]. It has additionally been shown in vivo that activation of Kupffer cells and sinusoidal endothelial cells upon acute endotoxemia is largely dependent on TLR-4 [29]. In line with this, livers from Fas wt animals subjected to ALF showed a 2-fold rise in TLR-4 expression [30]. The observation from our current study that in Gal/LPS-challenged Fas lpr mutant mice, TLR-4 expression is markedly suppressed strongly indicates that interruption of Fas ligation modulates TLR-4 signalling [30], thereby contributing to the prevention of liver injury.
In addition to TNFα and its receptors, which signal apoptosis via an intracellular suicide cascade, ligation of cell-membrane Fas also triggers apoptosis in many cell types, including hepatocytes [31]. Although Fas has been implicated in a variety of chronic liver diseases that are characterized by hepatocellular apoptosis [4, 13, 32–34], controversy exists regarding the contribution of Fas/FasL to inflammation-induced programmed cell death. Previous reports that neither Fas antigen–negative MRL/lpr nor FasL mutant mice were protected against Gal/LPS-induced hepatic injury [6, 8] are in contrast with the findings of the present study, in which Fas lpr mutation conferred significant protection, as shown by the diminution in the number of apoptotic hepatocytes and the decreased activation of the downstream effector caspase-3. Caspase-3 is reported to be essential for Fas-mediated apoptosis in vitro [35]. An in vivo study could further demonstrate that caspase-3 is required for initial events that occur in hepatocyte cell death following Fas ligation [36]. Along with cleaved caspase-3 positive hepatocytes, we demonstrated a Gal/LPS-induced increase in Fas and FasL levels by use of immunhistochemistry, RT-PCR, and Western blot analysis, which was far less pronounced when TNFα was blocked. These findings strongly indicate that TNFα-induced activation of the Fas/FasL pathway may be a critical factor in the amplification of hepatocyte apoptosis, because FasL-expressing hepatocytes can cause Fas-mediated suicide or fratricide [13, 33]. This view is further underlined by the inhibition of TNFα-induced apoptosis of HepG2 cells by a neutralizing anti-FasL antibody. Furthermore, we determined the extent of apoptosis in HepG2 cells which have been cultured at high density. The tight cell-cell-contact might increase the degree of apoptosis in comparison to cells cultured at lower density (data not shown). In addition, Fas positive hepatocytes were found in the direct neighbourhood of hepatocytes which were FasL positive, therefore underscoring the fratricidal cell death by Fas-FasL interaction. Although we could not completely eliminate the possibility that inflammatory cells induce Fas/FasL-mediated hepatocellular apoptosis, we would like to conclude that particularly fratricidal death by Fas-FasL interactions of neighbouring hepatocytes may actively contribute to ALF.
Recently, it was shown that Fas ligation not only promotes apoptosis but also stimulates proinflammatory events such as macrophage infiltration and leukocyte recruitment during liver injury [37]. Leukocyte infiltration has been shown to be not only the cause, but also the consequence of hepatocyte apoptosis [38]. Thus, the marked inhibition of hepatocellular apoptosis in the Fas lpr mice might be due—at least in part—to a reduction in inflammatory leukocyte infiltration.
In conclusion, TNFα hepatotoxicity in Gal/LPS-induced acute liver failure involves Fas/FasL-dependent cell death in either a fratricidal or suicidal manner. Thus, when developing anti-apoptotic strategies, hepatocytes should be considered to be active contributors to apoptotic cell death.
References
Khashab M, Tector AJ, Kwo PY (2007) Epidemiology of acute liver failure. Curr Gastroenterol Rep 9:66–73. doi:10.1007/s11894-008-0023-x
Schattenberg JM, Galle PR, Schuchmann M (2006) Apoptosis in liver disease. Liver Int 26:904–911. doi:10.1111/j.1478-3231.2006.01324.x
Faubion WA, Gores GJ (1999) Death receptors in liver biology and pathobiology. Hepatology 29:1–4. doi:10.1002/hep.510290101
Kondo T, Suda T, Fukuyama H et al (1997) Essential roles of the Fas ligand in the development of hepatitis. Nat Med 3:409–413. doi:10.1038/nm0497-409
Ksontini R, Colagiovanni DB, Josephs MD et al (1998) Disparate roles for TNF-alpha and Fas ligand in concanavalin A-induced hepatitis. J Immunol 160:4082–4089
Josephs MD, Bahjat FR, Fukuzuka K et al (2000) Lipopolysaccharide and d-galactosamine-induced hepatic injury is mediated by TNF-alpha and not by Fas ligand. Am J Physiol Regul Integr Comp Physiol 278:R1196–R1201
Ando K, Hiroishi K, Kaneko T et al (1997) Perforin, Fas/Fas ligand, and TNF-alpha pathways as specific and bystander killing mechanisms of hepatitis C virus-specific human CTL. J Immunol 158:5283–5291
Morikawa A, Sugiyama T, Kato Y et al (1996) Apoptotic cell death in the response of d-galactosamine-sensitized mice to lipopolysaccharide as an experimental endotoxic shock model. Infect Immun 64:734–738
Tagawa Y, Kakuta S, Iwakura Y (1998) Involvement of Fas/Fas ligand system-mediated apoptosis in the development of concanavalin A-induced hepatitis. Eur J Immunol 28:4105–4113. doi:10.1002/(SICI)1521-4141(199812)28:12<4105::AID-IMMU4105>3.0.CO;2-8
Seino K, Kayagaki N, Takeda K et al (1997) Contribution of Fas ligand to T cell-mediated hepatic injury in mice. Gastroenterology 113:1315–1322. doi:10.1053/gast.1997.v113.pm9322527
Freudenberg MA, Galanos C (1991) Tumor necrosis factor alpha mediates lethal activity of killed gram-negative and gram-positive bacteria in d-galactosamine-treated mice. Infect Immun 59:2110–2115
Guy CS, Wang J, Michalak TI (2006) Hepatocytes as cytotoxic effector cells can induce cell death by CD95 ligand-mediated pathway. Hepatology 43:1231–1240. doi:10.1002/hep.21201
Galle PR, Hofmann WJ, Walczak H et al (1995) Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J Exp Med 182:1223–1230. doi:10.1084/jem.182.5.1223
Philpott NJ, Turner AJ, Scopes J et al (1996) The use of 7-amino actinomycin D in identifying apoptosis: simplicity of use and broad spectrum of application compared with other techniques. Blood 87:2244–2251
Le Minh K, Klemm K, Abshagen K et al (2007) Attenuation of inflammation and apoptosis by pre- and posttreatment of darbepoetin-alpha in acute liver failure of mice. Am J Pathol 170:1954–1963. doi:10.2353/ajpath.2007.061056
Eipel C, Kidess E, Abshagen K et al (2007) Antileukoproteinase protects against hepatic inflammation, but not apoptosis in the response of d-galactosamine-sensitized mice to lipopolysaccharide. Br J Pharmacol 151:406–413. doi:10.1038/sj.bjp.0707230
Leist M, Gantner F, Bohlinger I et al (1995) Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol 146:1220–1234
Wooley PH, Dutcher J, Widmer MB et al (1993) Influence of a recombinant human soluble tumor necrosis factor receptor FC fusion protein on type II collagen-induced arthritis in mice. J Immunol 151:6602–6607
Schafer T, Scheuer C, Roemer K et al (2003) Inhibition of p53 protects liver tissue against endotoxin-induced apoptotic and necrotic cell death. FASEB J 17:660–667. doi:10.1096/fj.02-0774com
Galanos C, Freudenberg MA (1993) Mechanisms of endotoxin shock and endotoxin hypersensitivity. Immunobiology 187:346–356
Fecho K, Cohen PL (1998) Fas ligand (gld)- and Fas (lpr)-deficient mice do not show alterations in the extravasation or apoptosis of inflammatory neutrophils. J Leukoc Biol 64:373–383
Watanabe-Fukunaga R, Brannan CI, Copeland NG et al (1992) Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356:314–317. doi:10.1038/356314a0
Adachi M, Watanabe-Fukunaga R, Nagata S (1993) Aberrant transcription caused by the insertion of an early transposable element in an intron of the Fas antigen gene of lpr mice. Proc Natl Acad Sci USA 90:1756–1760. doi:10.1073/pnas.90.5.1756
Drappa J, Brot N, Elkon KB (1993) The Fas protein is expressed at high levels on CD4+CD8+ thymocytes and activated mature lymphocytes in normal mice but not in the lupus-prone strain, MRL lpr/lpr. Proc Natl Acad Sci USA 90:10340–10344. doi:10.1073/pnas.90.21.10340
Mohler KM, Sleath PR, Fitzner JN et al (1994) Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature 370:218–220. doi:10.1038/370218a0
Lehmann V, Freudenberg MA, Galanos C (1987) Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and d-galactosamine-treated mice. J Exp Med 165:657–663. doi:10.1084/jem.165.3.657
Tiegs G, Wolter M, Wendel A (1989) Tumor necrosis factor is a terminal mediator in galactosamine/endotoxin-induced hepatitis in mice. Biochem Pharmacol 38:627–631. doi:10.1016/0006-2952(89)90208-6
Su GL, Klein RD, Aminlari A et al (2000) Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 31:932–936. doi:10.1053/he.2000.5634
Chen LC, Gordon RE, Laskin JD et al (2007) Role of TLR-4 in liver macrophage and endothelial cell responsiveness during acute endotoxemia. Exp Mol Pathol 83:311–326. doi:10.1016/j.yexmp.007.08.015
Ma Y, Liu H, Tu-Rapp H et al (2004) Fas ligation on macrophages enhances IL-1R1-Toll-like receptor 4 signaling and promotes chronic inflammation. Nat Immunol 5:380–387. doi:10.1038/ni1054
Ogasawara J, Watanabe-Fukunaga R, Adachi M et al (1993) Lethal effect of the anti-Fas antibody in mice. Nature 364:806–809. doi:10.1038/364806a0
Guidotti LG, Ishikawa T, Hobbs MV et al (1996) Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4:25–36. doi:10.1016/S1074-7613(00)80295-2
Strand S, Hofmann WJ, Grambihler A et al (1998) Hepatic failure and liver cell damage in acute Wilson’s disease involve CD95 (APO-1/Fas) mediated apoptosis. Nat Med 4:588–593. doi:10.1038/nm0598-588
Roskams T, Libbrecht L, Van Damme B et al (2000) Fas and Fas ligand: strong co-expression in human hepatocytes surrounding hepatocellular carcinoma; can cancer induce suicide in peritumoural cells? J Pathol 191:150–153. doi:10.1002/(SICI)1096-9896(200006)191:2<150::AID-PATH612>3.0.CO;2-I
Woo M, Hakem R, Soengas MS et al (1998) Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev 12:806–819. doi:10.1101/gad.12.6.806
Woo M, Hakem A, Elia AJ et al (1999) In vivo evidence that caspase-3 is required for Fas-mediated apoptosis of hepatocytes. J Immunol 163:4909–4916
Fujiwara M, Suemoto H, Muragaki Y et al (2007) Fas-mediated upregulation of vascular endothelial growth factor and monocyte chemoattractant protein-1 expression in cultured dermal fibroblasts: role in the inflammatory response. J Dermatol 34:99–109. doi:10.1111/j.1346-8138.2006.00226.x
Eipel C, Bordel R, Nickels RM et al (2004) Impact of leukocytes and platelets in mediating hepatocyte apoptosis in a rat model of systemic endotoxemia. Am J Physiol Gastrointest Liver Physiol 286:G769–G776. doi:10.1152/ajpgi.00275.2003
Acknowledgements
The authors cordially thank Berit Blendow, Doris Butzlaff, Dorothea Frenz, Maren Nerowski, and Hartmut Stein (Institute for Experimental Surgery, University of Rostock, Germany) for excellent technical assistance, and Saleh Ibrahim (Immunogenetics Group, University of Rostock) for fruitful discussions and reviewing the manuscript. This study was supported by a grant from the Deutsche Forschungsgemeinschaft, Bonn Bad-Godesberg, Germany (Vo 450/8-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kuhla, A., Eipel, C., Siebert, N. et al. Hepatocellular apoptosis is mediated by TNFα-dependent Fas/FasLigand cytotoxicity in a murine model of acute liver failure. Apoptosis 13, 1427–1438 (2008). https://doi.org/10.1007/s10495-008-0269-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0269-7