
Abstract
Resveratrol (3,4′,5-trihydroxystilbene) is a phytochemical believed to be partly responsible for the cardioprotective effects of red wine due to its numerous biological activities. Here, we studied biochemical pathways underlying peroxynitrite-mediated apoptosis in endothelial cells and potential mechanisms responsible for resveratrol cytoprotective action. Peroxynitrite triggered endothelial cell apoptosis through caspases-8, -9 and -3 activation implying both mitochondrial and death receptor apoptotic pathways. Resveratrol was able to prevent peroxynitrite-induced caspases-3 and -9 activation, but not caspase-8 activation. Additionally, peroxynitrite increased intracellular levels of Bax without affecting those of Bcl-2, increasing consequently the Bax/Bcl-2 ratio. This ratio decreased when cells where pre-incubated with 10 and 50 μM resveratrol, mainly due to resveratrol ability per se to increase Bcl-2 intracellular levels without affecting Bax intracellular levels. These results propose an additional mechanism whereby resveratrol may exert its cardioprotective effects and suggest a key role for Bcl-2 in the resveratrol anti-apoptotic action, especially in disrupting peroxynitrite-triggered mitochondrial pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis is a multifaceted disorder where endothelial dysfunction plays a key role [1]. Endothelium is constituted by a single monolayer of endothelial cells that acts as a selective and permeable barrier and regulates multiple functions such as vascular tone, thrombosis, inflammation, platelet aggregation and vascular remodelling [2]. Therefore, any damage or stress in endothelial cells will disturb endothelium functions leading to its dysfunction. On the other hand, apoptosis of arterial endothelial cells is a prominent feature of atherosclerotic plaques which contributes to its instability and amplifies endothelial dysfunction, and subsequently atherosclerosis by inducing a dramatic inflammatory response [3]. Several evidences in literature point to the association between oxidative, nitrosative/nitrative stress and endothelial dysfunction, as atherosclerotic lesions occur predominantly in areas with disturbed blood flow where an increase in superoxide anion (O •−2 ) generation and a decrease in nitric oxide (NO) are observed [4]. This is of major importance since an overproduction of O •−2 by several enzymes, such as NAD(P)H oxidase, xanthine oxidase and uncoupled NOS, will counteract beneficial effects of NO on the vasculature by oxidizing it to the highly reactive species peroxynitrite (ONOO-) [5]. In fact, 3-nitrotyrosine, a biological marker of peroxynitrite generation, increases in human atherosclerotic tissue [6, 7]. Peroxynitrite is a powerful oxidant and a nitrating species able to react with and to modify a wide range of biomolecules, including DNA, proteins and lipids, leading to the dysfunction of critical cellular processes, disruption of cell signalling pathways, and the induction of cell death through both apoptosis and necrosis [8–10].
In vitro studies have shown that peroxynitrite could induce endothelial cell death either by an apoptotic or necrotic programmed pathway, depending on peroxynitrite cell exposure conditions and concentrations. Szabó and collaborators have reported a non passive necrotic cell death initiated by DNA oxidative injury and subsequent activation of the nuclear enzyme poly (ADP-ribose) polymerase-1 followed by depletion of NAD+ and ATP cellular pools [11, 12]. On the contrary, we and others have observed morphological changes in endothelial cells upon exposure to peroxynitrite consistent with apoptosis, but the apoptotic machinery remains poorly understood [13, 14].
Resveratrol (3,4′,5-trihydroxystilbene), a phytoalexin synthesized in several plants has been a focus of attention in the cardiovascular research field, since it has been detected in red wine and pointed to be responsible for the cardioprotective effects observed in moderate wine drinkers [15]. Afterwards, research has reported a myriad of resveratrol biological activities, including antioxidant, anti-inflammatory, anti-platelet aggregation and anti-proliferative activities which improve not only endothelial function but also the cardiovascular system in general, supporting a role for resveratrol as a cardioprotective agent [16–19]. Previously, we have shown that resveratrol, at a high concentration (50 μM), was able to prevent endothelial cell death through an indirect antioxidant activity by increasing the intracellular glutathione (GSH) pools [14]. However, other cellular mechanisms should account for the cytoprotective effects of resveratrol because at a low concentration (10 μM), it was able to counteract peroxynitrite deleterious effects on endothelial cells without increasing the cellular glutathione concentration. So, in this work, we aimed to investigate biochemical pathways underlying peroxynitrite-induced apoptosis and to explore new resveratrol cytoprotective mechanisms against cell injury elicited by peroxynitrite.
The data reported in this study indicate that peroxynitrite induces bovine aortic endothelial cells (BAEC) apoptosis by triggering both mitochondrial and death receptor pathway and show, for the first time, that resveratrol prevents peroxynitrite cell deleterious effects by disrupting the mitochondrial apoptotic pathway most likely via increase in the anti-apoptotic Bcl-2 protein levels.
Materials and methods
Materials
General laboratory chemicals and some specific ones, namely, collagenase, gelatin, streptomycin/penicillin, MTT, Hoechst 33258, 4′6-diamidino-2-phenylindole (DAPI) and protease inhibitor cocktail were obtained from Sigma Chemicals (St. Louis, MO, USA). For cell culture, Dulbecco’s modified Eagle’s medium (DMEM), trypsin 0.25%, fungizone, fetal bovine serum (FBS) and phosphate-buffered saline (PBS) pH 7.4, were purchased from Gibco-Invitrogen. Resveratrol was from Extrasynthèse (Genay, France). Caspases substrates DEVD-7-AMC, IEPD-7-AMC and LEDH-7-AFC were purchased from Bachem (Bubendorf, Switzerland). Primary specific mouse monoclonal antibody to Bax and anti-mouse IgG secondary antibody were obtained from Abcam (Cambridge, UK); primary specific mouse monoclonal antibody to Bcl-2 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), primary specific mouse monoclonal antibody to β-actin was purchase from Sigma (St. Louis, MO, USA).
Primary cultures of bovine aortic endothelial cells
Bovine aortic endothelial cells were isolated from thoracic aorta after digestion with collagenase (2 mg/ml). Subsequently, cells were plated on gelatin-coated tissue culture plastic and maintained in DMEM supplemented with 10% heat inactivated fetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml) and fungizone (50 ng/ml) at 37°C under an humidified atmosphere of 5% CO2. Endothelial cells were identified by their cobblestone morphology and uptake of fluorescently labelled acetylated LDL. Cells were subcultured at confluences and used between the fourth and the seventh passage. Prior to the experiments, cells at 80% of confluence were starved in serum-free medium for 24 h.
Peroxynitrite synthesis
Peroxynitrite was synthesized by using a quenched flow reactor, as previously described [20]. Briefly, an aqueous solution of 0.6 M NaNO2 was mixed rapidly with an equal volume of 0.7 M H2O2 containing 0.6 M HCl and immediately quenched with the same volume of 1.5 M NaOH. The solution was then frozen at −20°C for approximately 24 h. At this temperature, peroxynitrite concentrates into a dark yellow top layer (freeze fractionation), which was removed and passed through a short column of MnO2 for residual H2O2 remotion according to Uppu and Pryor [21]. Then, it was stored at −80°C under N2 atmosphere. Before use, ONOO− was always quantified from the absorbance at 302 nm in 1 N NaOH (ɛ = 1,670 cm−1M−1).
Cell treatment
Cell death was triggered by an oxidative stress induced by peroxynitrite. For this purpose, cells were washed once with PBS and equilibrated with PBS pH 7.4 for 5 min. Subsequently, 500 μM ONOO− (final concentration), previously diluted in 10 mM NaOH to the required concentration, was delivered as a single bolus against one side of the dish while rapidly swirling the medium to ensure optimal exposure of the cells to ONOO− before decomposition. Cells were exposed to ONOO− for 10 min, after which they were washed and replaced in culture medium for the indicated times that could reach 6 h maximum. No pH shift was observed during treatment with ONOO−. Peroxynitrite-induced oxidative stress was preformed rather in PBS than DMEM in order to avoid interfering reactions of ONOO− with media constituents. Same volumes of 10 mM NaOH (vehicle control) or decomposed ONOO− (ONOO− was decomposed in PBS) were used as controls.
When required, cells were pre-incubated with resveratrol for 14 h. At the end of this time, cells were washed and incubated with ONOO− as described above. In these conditions resveratrol was not present in the incubation medium during and after ONOO− treatment.
Cell viability assay
Cell viability was evaluated by the MTT test [22]. At the indicated times cells were washed twice with PBS and subsequently incubated with MTT (5 mg/ml in PBS) for 2 h in the dark at 37°C. Then, the MTT in each well was taken out carefully and formazan crystals were redissolved in DMSO. Absorbance of this solution was read at 530 nm in a Perkin–Elmer spectrophotometer. Results were expressed as percentage of MTT reduction, assuming that the absorbance of control cells was 100%.
Nuclear morphology by fluorescent staining
Cells were fixed with 4% (w/v) paraformaldehyde in phosphate buffered saline (PBS) (pH 7.4) for 15 min at room temperature and subsequently stained with Hoechst dye 33258 (5 μg/ml) for 15 min, washed with PBS and mounted by using PBS/glycerol (3:1, v/v). Then, cells were examined by fluorescence microscopy, with a Nikon fluorescence microscope. Stained nuclei were scored according to the condensation and staining characteristics of chromatin. Apoptotic nuclei were identified by condensed chromatin and nuclear fragmentation and were counted in at least six random microscopic fields (400×) per sample. Only cells that remained attached to the plates were observed. Mean values were expressed as the percentage of apoptotic nuclei.
Quantification of DNA fragmentation
DNA fragmentation was evaluated as previously described by McConkey et al. [23]. In brief, cells were recovered, ressuspended in ice-cold lysis buffer [5 mM Tris pH 8.0, 0.5% (v/v) Triton X-100, 20 mM EDTA] for 15 min and ultracentifuged for 20 min at 27,000g to separate intact chromatin (pellet) from DNA cleavage products (supernatant). The pellet ressuspended in 1 ml of 10 mM Tris–HCl, pH 8.0 buffer containing 1 mM EDTA and the supernatant were assayed for DNA content by the fluorimetric DAPI procedure according to Kapuscinski and Skooczylas [24]. Mean values of DNA fragmentation were expressed as the percentage of total DNA.
Caspase activity assays
Caspase activation was evaluated in cytosolic protein extracts. Briefly, at the end of the incubation times, cells were recovered and ressuspended in ice-cold cell lysis buffer [25 mM HEPES pH 7.4, 0.1% (w/v) CHAPS, 1 mM Na-EDTA, 1 mM Na-EGTA, 2 mM MgCl2, 100 μM PMSF, 2 mM DTT, 1/100 cocktail protease inhibitors (v/v)] for 20 min. Subsequently, cells were frozen/thawed five times (liquid nitrogen/37°C) and centrifuged at 16,000g for 10 min at 4°C. The supernatant was then collected and stored at −80°C. Protein concentration was determined by using the Bio-Rad protein assay kit according to the manufacture’s specifications (Bio-Rad, USA). The enzymatic caspases activities were determined by using substrates containing specific cleavage sites, linked to a fluorochrome. Equal amounts of protein (20–25 μg) were incubated in an assay buffer [HEPES pH 7.4, 10% (w/v) sucrose, 0.1% (w/v) CHAPS, 10 mM DTT] with 100 μM of fluorogenic peptide substrates, DEVD-AMC (caspase-3 activity) IEPD-AMC (caspase-8 activity) and LEDH-AFC (caspase-9 activity), for 2 h at 37°C. Subsequently, release of AMC or AFC was determined by fluorescence measurement in a Perkin–Elmer LS50 spectrometer at an λexc = 390 nm and λem = 475 (slits with 5 nm) for AMC, and λexc = 395 nm and λem = 495 nm (slits with 5 nm) for AFC. Caspases activities were calculated as the increase above the control for equal protein loaded.
Measurement of mitochondrial membrane potential
Cells were incubated with 500 nM tetramethylrhodamine methyl ester (TMRM) 30 min before the end of the experiments [25]. Subsequently, cells were washed, harvested and ressuspended in DMEM without phenol red and fluorescence intensity was measured in a Perkin–Elmer LS50 spectrometer at an λexc = 550 nm and λem = 572 nm (slits with 10 nm). Under these conditions there was no evidence of TMRM cytotoxicity and TMRM was used in a non-quenched mode as observed by the absence of increase in fluorescence intensity after addition of FCCP to cells suspension in the cuvette. To confirm the measurement of mitochondrial membrane potential under these conditions, cells were pre-incubated with 2 μM FCCP for 10 min and as expected a decrease in fluorescence intensity was observed. The results were normalized to the control fluorescence intensity using the same number of cells (1.5 × 106/ml).
Immunoblot
Total protein cell extracts were obtained after cell suspension lysis in an ice-cold lysis buffer [50 mM Hepes pH 7.4, 150 mM NaCl, 2 mM EDTA, 10% (w/v) glycerol, 0.5% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100, 1 mM PMSF, 1/100 (v/v) proteases cocktail inhibitor] for 20 min. Lysates were subsequently centrifuged at 16,000g for 10 min at 4°C and supernatants were then collected and stored at −80°C. Protein concentration was determined by using the Bio-Rad protein assay kit, according to the manufacturer’s specifications. Equal amount of proteins (25–40 μg) were analyzed by electrophoresis on a 12 or 15% SDS-polyacrylamide gel and were blotted to polyvinylidene difluoride (PVDF) membranes (Amersham, Buckinghamshire, UK) for 2 h at 200 mA. To avoid non-specific binding, membranes were blocked for 1 h at room temperature with 5% (w/v) non-fat dried milk in TBS-T buffer [25 mM Tris–HCl pH 7.6, 150 mM NaCl, 0.1% (v/v) Tween 20]. Membranes were then incubated overnight at 4°C with primary mouse monoclonal anti-Bax (dilution 1:10,000) and anti-Bcl-2 (dilution 1:300) antibodies. After five washings with TBS-T, membranes were incubated with phosphatase alkaline-labelled secondary antibody anti-mouse (dilution 1:20,000) for 1 h at room temperature. Membranes were washed again five times with TBS-T and transferred proteins were incubated with enhanced chemifluorescence reagent (Amersham, UK), according to the manufacturer instruction and visualized by the VersaDoc Imaging System (BioRad, Hércules, CA, USA). β-Actin was used as a control for protein loading.
Statistical analysis
All data were expressed as mean ± SEM of at least three independent assays, each one in duplicate. Significance between groups was assessed by one-way analysis of variance (ANOVA) followed by Tukey’s or Dunnet’s (time course assays) post hoc tests. A value of P lower than 0.05 was considered statistically significant.
Results
Resveratrol prevented caspase-3 and -9 but not caspase-8 activation induced by peroxynitrite
It has been well established that at the biochemical level, apoptosis may be mediated through activation of initiator and effector caspases, namely caspases-8 and -9, and caspase-3, respectively [26]. Thus, we investigated the putative role of caspases in resveratrol protective effects against peroxynitrite-mediated cell death.
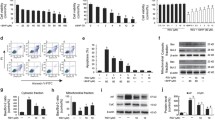
Nuclear morphological changes of BAEC assessed after nuclear staining with Hoechst and DNA fragmentation assessed by the DAPI quantitative procedure corroborated our previous results that a 14 h pre-incubation of BAEC with 10 or 50 μM resveratrol rescued cells from apoptosis induced by 500 μM peroxynitrite (Fig. 1a). Indeed, the significant level of apoptosis (about 25%) observed 6 h after cells treatment with 500 μM peroxynitrite was efficiently decreased by resveratrol, in a concentration-dependent manner. Of note, resveratrol was not present in the medium during or after peroxynitrite-treatment and none of the tested concentrations was cytotoxic to endothelial cells up to 48 h as indicated by the MTT test (Fig. 1b).

Resveratrol prevents peroxynitrite-mediated apoptosis in endothelial cells. Confluent bovine aortic endothelial cells maintained in a medium free of serum were pre-incubated with 10 and 50 μM resveratrol for 14 h. After this time, cells were washed once with PBS and equilibrated with PBS pH 7.4 for 5 min. Then, cells were exposed to 500 μM peroxynitrite in 1 ml/well of PBS for 10 min. Subsequently, PBS was removed and replaced by fresh serum-free medium and incubated at 37°C for 6 h. Morphological apoptotic changes and the DNA fragmentation were then assessed after nuclei staining with Hoechst 33258 and by the DAPI fluorimetric procedure after ultracentrifugation of DNA, respectively. Control refers to the experiment in similar conditions without peroxynitrite and resveratrol. (a) Results of apoptosis quantification were expressed as % of apoptotic cells or % of DNA fragmentation relatively to the total cell population or the total DNA, respectively. (b) Time course of cellular viability of BAEC treated with different concentrations of resveratrol and expressed as % of MTT reduction. Values are mean ± SEM of five experiments, each one assayed in duplicate. ***P < 0.001 versus control, ### P < 0.001 versus peroxynitrite without resveratrol
Subsequently, we investigated the biochemical pathways underlying peroxynitrite-induced apoptosis by evaluating the time courses of caspases-3, -8 and -9 activation. At shorter post-stimulation times (0–1 h), peroxynitrite reduced, yet in a non-significant way, all tested caspases activities to about half the control activity (Fig. 2). However, this initial caspases inactivation by peroxynitrite was followed by a significant increase in a time dependent-manner. Increases of about 2.3 times those of controls in caspases-3 (P < 0.05) and -8 (P < 0.01) activities and of about 2-fold in caspase-9 (P < 0.05) activity were observed 3 h after peroxynitrite-treatment. Throughout the time course, caspases activities increased significantly reaching values about 3.8-fold (P < 0.01), 3.1-fold (P < 0.01) and 2.4-fold (P < 0.01) higher than control for caspases-3, -8 and -9 respectively, 6 h after peroxynitrite-treatment. Once established the involvement of caspases-3, -8 and -9 in peroxynitrite-mediated BAEC apoptosis we investigated whether resveratrol could modulate or not such caspases activation. Resveratrol inhibited peroxynitrite-induced caspase-3 activity, in a concentration-dependent manner (Fig. 3a). Whereas 10 μM resveratrol reduced by about 32% the 4.6-fold-increase in caspase-3 activity observed 3 h after peroxynitrite treatment, a significantly higher reduction (51%) was achieved in cells pre-treated with 50 μM resveratrol. In contrast, none of the tested resveratrol concentrations prevented significantly the 3.6-fold-increase in caspase-8 activity 3 h after peroxynitrite treatment (Fig. 3b). The lowest tested resveratrol concentration exhibited no counteracting effects on peroxynitrite-induced caspase-8 activity and although, the highest resveratrol concentration tested decreased it, such decrease is not statistically significant. Finally, both resveratrol concentrations under study thwarted efficiently caspase-9 activity triggered by peroxynitrite, as shown in Fig. 3c. The 2.8-fold-increase in caspase-9 activity, observed 3 h after cells’ treatment with peroxynitrite was reduced by 34% or by 52% after pre-incubating cells with 10 or 50 μM resveratrol, respectively. As caspase-9 and caspase-8 are well-known initiator caspases of the mitochondrial and death receptor apoptotic pathways, respectively, these results suggest that resveratrol inhibits peroxynitrite-induced endothelial cell apoptosis, preferentially by disrupting the mitochondrial apoptotic pathway than the death receptor pathway triggered by peroxynitrite [27].

Apoptosis induced by peroxynitrite is mediated by caspases-3, -8 and -9. Confluent BAEC were treated with peroxynitrite as described in the legend of Fig. 1 and caspases activities were quantified at different times (0–360 min) after cells treatment. Caspases activities were measured with fluoregenic substrates, as described in “Materials and methods”. Values are mean ± SEM of 4–6 experiments each one assayed in duplicate. *P < 0.05 versus untreated control cells (C), **P < 0.01 versus C

Resveratrol inhibits caspases-3 and -9 but not caspase-8 activation induced by peroxynitrite. Confluent BAEC were treated with peroxynitrite and resveratrol as described in the legend of Fig. 1. Following a 14 h pre-incubation of cells with 10 or 50 μM resveratrol, cells were treated with 500 μM peroxynitrite for 10 min and 3 h after this treatment, caspases activities were evaluated as described in “Materials and methods”. Activities of (a) caspase-3, (b) caspase-8 and (c) caspase-9. Values are mean ± SEM of 5 (a and b) or 6 (c) experiments, each one assayed in duplicate. *P < 0.05 versus control, ***P < 0.001 versus control, # P < 0.05 versus peroxynitrite, ## P < 0.01 versus peroxynitrite, ### P < 0.001 versus peroxynitrite
Resveratrol did not prevent mitochondrial membrane potential dissipation elicited by peroxynitrite
Mitochondrial outer membrane (MOM) permeabilization and subsequent release of soluble intermembrane space proteins, such as cytochrome c, into the cytosol where it is responsible for caspase-9 activation, are key events in the apoptotic mitochondrial pathway [28]. One of the mechanisms recognized to contribute to MOM permeabilization is the mitochondrial inner membrane transitory permeabilization through permeability transition pore (PTP) opening [29]. Therefore, we evaluated peroxynitrite effects on endothelial cells’ mitochondrial membrane potential. Peroxynitrite elicited a significant decrease in mitochondrial membrane potential that in our model reached a maximum of about 60%, 1 h after cells’ treatment and the decrease remained stable until the end of the time course study (6 h) (Fig. 4). Then, we investigated resveratrol effects on such induced mitochondrial membrane potential dissipation. None of the resveratrol concentrations tested protected cells from peroxynitrite-promoted mitochondrial membrane potential dissipation (Fig. 5), suggesting that the protective effects of resveratrol at the mitochondria level do not occur by resveratrol interference with the PTP, but preferentially through another mechanism. Resveratrol per se has no effect on the mitochondrial membrane potential (data not shown).

Peroxynitrite induces mitochondrial membrane potential dissipation. Mitochondrial membrane potential was measured at different times (30–360 min) after cells treatment with 500 μM peroxynitrite, as described in “Materials and methods”. Values are mean ± SEM of 4 experiments each one assayed in duplicate. *P < 0.05 versus control (C), **P < 0.01 versus C

Resveratrol does not inhibit the mitochondrial membrane potential dissipation induced by peroxynitrite. Mitochondrial membrane potential was measured 1 h after peroxynitrite treatment in cells pre-incubated for 14 h with 10 or 50 μM resveratrol, as described in “Materials and methods”. Values are mean ± SEM of 4 experiments each one assayed in duplicate. **P < 0.01 versus control
Resveratrol increased Bcl-2 protein intracellular levels reducing the peroxynitrite-induced Bax/Bcl-2 ratio increase
Alternatively to the PTP, it has been proposed that Bcl-2 family proteins, important regulators of apoptosis in mammalian cells, are implicated in the MOM permeabilization [30]. Bcl-2 family proteins are divided into anti-apoptotic proteins (Bcl-2, Bcl-XL, etc.) and pro-apoptotic proteins (Bax, Bak, etc.) that have been demonstrated to display opposite activities in mitochondria. Consequently, the balance between pro- and anti-apoptotic proteins is vital for the control of cell survival. Thus, we investigated if peroxynitrite would be able to modify the intracellular levels of Bcl-2 family members, namely, Bax and Bcl-2. As shown in Fig. 6a, 30 min after cells treatment with peroxynitrite, a 2.1-fold increase in Bax protein intracellular levels was observed. This increase reached a maximal value 1 h post-stimulation time but was not sustained during the entire experiment time course. After 1 h, Bax protein levels decreased to values relatively constants up to the end of the experiment. Conversely, Bcl-2 protein levels were not significantly affected by peroxynitrite throughout the time course study, indicating that this species might initiate endothelial cell death by varying the balance between Bax and Bcl-2 (Fig. 6b). In fact, an increase in Bax levels without changes in Bcl-2 levels leads to a raise in Bax/Bcl-2 ratio, disrupting the intracellular balance between pro-apoptotic and anti-apoptotic proteins. Since, Bax and Bcl-2 may be key elements in peroxynitrite-induced apoptosis, we explored resveratrol ability to modulate intracellular levels of these proteins and consequently to hamper peroxynitrite effects.

Peroxynitrite increases Bax intracellular levels without affecting Bcl-2 intracellular levels. BAEC were treated with 500 μM peroxynitrite as described in the legend of Fig. 1 and at the indicated times (0–360 min) total protein extracts were analyzed by immunoblot with specific antibodies against (a) Bax and (b) Bcl-2, as described in “Materials and methods”. Representative blots and densitometric quantifications of four independent experiments are shown. Results are normalized to endogenous β-actin and expressed as mean ± SEM arbitrary units of at least 4 independent experiments. *P < 0.05 versus control (C), **P < 0.01 versus C
Resveratrol per se did not affect intracellular levels of Bax, but and unexpectedly, it exhibited a tendency to reduce, although in a non significant mode, the peroxynitrite-induced increase in Bax intracellular levels (Fig. 7a). On the other hand, resveratrol per se induced a significant and dose-dependent increase in Bcl-2 intracellular levels. Indeed, increases of 1.44 and 1.78-fold in Bcl-2 intracellular levels were reached in the presence of 10 and 50 μM resveratrol, respectively (Fig. 7b). Moreover, Bcl-2 cellular levels also increased up to 1.38-fold in cells pretreated with 10 or 50 μM resveratrol for 14 h and subsequently stimulated with peroxynitrite (Fig. 7b). Therefore, as shown in Fig. 7c, Bax/Bcl-2 ratio increased up to 2-fold, 1 h after cells peroxynitrite treatment. This effect was completely abolished by both resveratrol concentrations tested, probably, as a consequence of the decrease in Bax/Bcl-2 ratio that occurred in the presence of resveratrol alone (Fig. 7c). Altogether, these results suggest that both resveratrol concentrations tested may interfere with the mitochondrial apoptotic pathway triggered by peroxynitrite via modulation of intracellular levels of Bcl-2 family proteins.

Resveratrol increases Bcl-2 intracellular levels, reducing the peroxynitrite-induced Bax/Bcl-2 ratio increase. Total proteins were extracted from cells incubated with 10 and 50 μM resveratrol for 14 h with no further treatment or post-treated with 500 μM peroxynitrite for 10 min, as described in the legend of Fig. 1 and maintained for 1 h in serum-free medium at 37°C. Total protein extracts were then analyzed as described in legend of Fig. 6. Representative blots and densitometric quantifications of (a) Bax and (b) Bcl-2 and additionally (c) Bax/Bcl ratio, of at least four independent experiments are shown. Results are normalized to endogenous β-actin and expressed as mean ± SEM arbitrary units of at least 4 independent experiments. *P < 0.05 versus control (C), **P < 0.01 versus C, ## P < 0.01 versus ONOO−
Discussion
The biochemical mechanisms underlying peroxynitrite-mediated apoptosis and cytoprotection afforded by resveratrol are not yet fully understood. We have previously reported that resveratrol prevents cell death triggered by peroxynitrite by up-regulating glutathione intracellular levels [14]. Now, we provide valuable new insights into the biochemical cytoprotective mechanisms for resveratrol. In fact, the results presented in this study point out biochemical pathways underlying apoptosis triggered by peroxynitrite in endothelial cells and show that resveratrol inhibits the mitochondrial apoptotic pathway.
A growing body of evidences has demonstrated that depending on the cell type and concentration used, peroxynitrite elicits different patterns of cell death, such as caspases-dependent or -independent apoptosis and necrosis [31–33]. Our data on caspases activation reported here support, for the first time, an apoptotic pathway for peroxynitrite toxicity in endothelial cells. Caspases are key elements of the machinery implicated in apoptosis, and can be activated extrinsically through death receptors or intrinsically through apoptogenic factors released from mitochondria [27]. Caspases-8 and -9, initiator caspases of the death receptors and of the mitochondrial pathway, respectively, are able to activate downstream effector caspases, like caspase-3 [26]. The activation of all three caspases after treatment of the cells with peroxynitrite, observed in this study, indicates that both death receptor and mitochondrial pathways are triggered by such reactive species. In fact, caspases activation is only observed after 2 h post-stimulation time, i.e., when peroxynitrite is no longer present, and increases until the end of the experiment. On the contrary, for shorter post-stimulation times, we observed caspases inactivation, probably due to a direct reaction between the oxidant and the Cys residue present in the active site of the enzymes, since peroxynitrite reaction with thiol groups is one of the fastest and most important reactions described for peroxynitrite [34].
Mitochondrial outer membrane (MOM) permeabilization is an obligatory event for the release of apoptogenic factors, including cytochrome c that, once in the cytosol, binds to Apaf-1 leading to the apoptosome building, a platform that in turn recruits and activates pro-caspase-9 [29]. Extensive evidence has been accumulated demonstrating that the mitochondrial outer membrane permeabilization can be mediated via permeability transition pore (PTP) or via Bcl-2 family proteins [30, 35, 36]. Our results suggest that peroxynitrite induces MOM permeabilization by increasing intracellular levels of Bax, without affecting Bcl-2 intracellular levels. Bax is a pro-apoptotic member of the Bcl-2 family that has been implicated in MOM permeabilization, via formation of proteic pores, lipidic pores or via interaction with pre-existing mitochondrial pores, like PTP [30]. Moreover, Bax cellular activity is antagonized through the formation of heterodimmers with anti-apoptotic proteins, more specifically with Bcl-2. In this way, the ratio Bax/Bcl-2 determines the fate of the cell survival and cell death type.
The role of PTP in MOM permeabilization, and the subsequent peroxynitrite-elicited caspase-9 activation in endothelial cells, has not been totally clarified. PTP opening is associated with mitochondrial membrane potential dissipation, mitochondrial swelling and MOM rupture [35, 36]. In our model and in agreement with several other reports, peroxynitrite induced a rapid and significant decrease in the mitochondrial membrane potential [37–40], which can be ascribed to PTP and consequently to the mitochondrial permeability transition event. In fact, peroxynitrite has been largely described as a mitochondrial permeability transition (MPT) inducer in isolated mitochondria, an effect that has been associated to peroxynitrite ability to oxidize Cys residue thiol groups of adenine nucleotide translocase (ANT), one of the PTP constituents [41]. However, the dissipation of the mitochondrial membrane potential can also be the result of either the respiratory chain inhibition, considering that several chain components are inhibited by peroxynitrite or a consequence of a severe energetic depletion [37, 42, 43]. Additionally, in the last few years, MPT has been observed during the onset of necrosis and therefore appears to be a non-exclusive process of apoptosis [44].
On the other hand, the absence of resveratrol effects on the decrease in peroxynitrite-elicited mitochondrial membrane potential, strengthens the idea that the mitochondrial membrane potential dissipation is better related to necrosis than to apoptosis. Thus, it is reasonable to consider that in our experimental settings mitochondrial membrane potential dissipation observed is the sum of different effects of peroxynitrite, with different consequences in mitochondria.
Although the concentration of peroxynitrite used in this work seems to be relatively high, the net exposure of cells to peroxynitrite is much lower than 500 μM as it has a half life less than 1 s at 37°C, pH 7.4 [45]. Moreover, in vivo peroxynitrite formation has been estimated to be as high as 50–100 μM to 1 mM [46].
The increase in Bcl-2 levels can be an important mechanism whereby resveratrol protects cells from peroxynitrite-triggered apoptosis. In other studies, Bcl-2 overexpression has been shown to prevent apoptosis but not necrosis elicited by peroxynitrite [37, 40, 41]. Indeed, while Bcl-2 inhibits, e.g., caspases activation induced by peroxynitrite, it has no effect on mitochondrial membrane potential dissipation associated to non-passive necrosis [37]. So, resveratrol, in a similar way to Bcl-2, and probably by raising Bcl-2 intracellular levels, probably protects cells from apoptosis but not from necrosis triggered by peroxynitrite.
The mechanisms underlying resveratrol ability to modulate Bcl-2 intracellular levels in endothelial cells are unclear. At least two possible mechanisms, that can act concurrently, may account for this resveratrol effect: one, based on its antioxidant properties that influence cellular redox state and consequently cellular signaling pathways and redox-sensitive transcription factors; another, based on its non-antioxidant properties. We presume that the preferential mechanism by which resveratrol modulates Bcl-2 cellular levels will vary with its concentration. Indeed, resveratrol, at the highest concentration tested in this study, not only exhibits a higher capacity to scavenge free radicals but also improves the redox cellular state, by increasing enzymatic and non-enzymatic antioxidant cellular system, such as SOD, catalase, glutathione redutase and more specifically GSH in BAEC [14, 47]. Thus, it is more likely that in our experimental model, 50 μM resveratrol modulates Bcl-2 levels by affecting the redox cellular state, whereas 10 μM resveratrol has not a significant role on that state and consequently on the redox-sensitive mechanisms. Actually, it has been recently reported that pharmacological preconditioning of rat hearts with 10 μM resveratrol increases Bcl-2 levels through adenosine A1 and A3 receptors activation, the former transmitting a survival signal through PI3K-Akt-Bcl-2 signaling pathway and the latter through CREB-dependent Bcl-2 pathway in addition to an Akt-Bcl-2 pathway [48]. Also, it has been shown that resveratrol modulates estrogen receptor-α-associated PI3K activity in a biphasic way, inducing activation at 10 μM and inactivation at higher concentrations [49]. The mechanism by which resveratrol modulates Bcl-2 cellular levels will need to be investigated in more details in future work.
While most data in the literature support a pro-apoptotic role for resveratrol, several studies report anti-apoptotic actions as observed in this work [50–53]. Although the reason for these discrepancies is not completely clear, both resveratrol concentration and cell type used may be relevant factors. Concerning cell type, resveratrol effects on cell proliferation and viability appears to be dependent on the cellular growth rates, i.e., the higher the proliferation rate the higher the sensitivity to resveratrol toxic effects. This idea is supported by the observation that freshly isolated normal human cells, with low passage number, are less sensitive to the pro-apoptotic effects of resveratrol than immortalized malignant or not malignant cell lines characterized by higher growth rates [54, 55]. It has been suggested that DNA synthesis could be the link between growth rates and cell sensitivity to resveratrol due to its ability to inhibit crucial enzymes involved in DNA synthesis, namely ribonucleotide reductase and DNA polymerase [56, 57].
In summary, this study confirms a biochemical apoptotic pathway for peroxynitrite-triggered endothelial cell death and expands our knowledge about the cellular mechanism implicated in resveratrol cytoprotective activities. Our results showed that resveratrol, at concentrations so low as 10 μM, inhibited biochemical and cellular mechanisms underlying apoptosis elicited by peroxynitrite and that resveratrol preferentially disrupted the mitochondrial apoptotic pathway than the death receptor apoptotic pathway, through modulation of Bcl-2 intracellular levels, considerably decreasing the Bax/Bcl-2 ratio. Furthermore, this study highlights the idea that resveratrol cardioprotective effects go far beyond its direct antioxidant activity and that its cellular signaling and molecular effects have a putative role to play.
Abbreviations
- NOS:
-
Nitric oxide synthase
- PARP-1:
-
Poly(ADP-ribose) polymerase-1
- BAEC:
-
Bovine aortic endothelial cells
- TMRM:
-
Tetramethylrhodamine methyl ester
- MOM:
-
Mitochondrial outer membrane
- PTP:
-
Permeability transition pore
- MPT:
-
Mitochondrial permeability transition
- GSH:
-
Glutathione
- ANT:
-
Mitochondrial inner membrane protein adenine nucleotide translocase
- CREB:
-
cAMP response element-binding protein
References
Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352:1685–1695. doi:10.1056/NEJMra043430
Ross R (1999) Atherosclerosis—an inflammatory disease. N Engl J Med 340:115–126. doi:10.1056/NEJM199901143400207
Choy JC, Granville DJ, Hunt DW, McManus BM (2001) Endothelial cell apoptosis: biochemical characteristics and potential implications for atherosclerosis. J Mol Cell Cardiol 33:1673–1690. doi:10.1006/jmcc.2001.1419
Cooke JP (2003) Flow, NO, and atherogenesis. Proc Natl Acad Sci USA 100:768–770. doi:10.1073/pnas.0430082100
Cai H, Harrison DG (2000) Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87:840–844
Beckmann JS, Ye YZ, Anderson PG, Chen J, Accavitti MA, Tarpey MM et al (1994) Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler 375:81–88
Leeuwenburgh C, Hardy MM, Hazen SL et al (1997) Reactive nitrogen intermediates promote low density lipoprotein oxidation in human atherosclerotic intima. J Biol Chem 272:1433–1436. doi:10.1074/jbc.272.3.1433
Radi R, Beckman JS, Bush KM, Freeman BA (1991) Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys 288:481–487. doi:10.1016/0003-9861(91)90224-7
Salgo MG, Stone K, Squadrito GL, Battista JR, Pryor WA (1995) Peroxynitrite causes DNA nicks in plasmid pBR322. Biochem Biophys Res Commun 210:1025–1030. doi:10.1006/bbrc.1995.1759
Ischiropoulos H, al Mehdi AB (1995) Peroxynitrite-mediated oxidative protein modifications. FEBS Lett 364:279–282. doi:10.1016/0014-5793(95)00307-U
Szabo C, Cuzzocrea S, Zingarelli B, O’Connor M, Salzman AL (1997) Endothelial dysfunction in a rat model of endotoxic shock. Importance of the activation of poly (ADP-ribose) synthetase by peroxynitrite. J Clin Invest 100:723–735. doi:10.1172/JCI119585
Virag L, Szabo E, Gergely P, Szabo C (2003) Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett 140-141:113–124. doi:10.1016/S0378-4274(02)00508-8
Foresti R, Sarathchandra P, Clark JE, Green CJ, Motterlini R (1999) Peroxynitrite induces haem oxygenase-1 in vascular endothelial cells: a link to apoptosis. Biochem J 339:729–736. doi:10.1042/0264-6021:3390729
Brito PM, Mariano A, Almeida LM, Dinis TC (2006) Resveratrol affords protection against peroxynitrite-mediated endothelial cell death: a role for intracellular glutathione. Chem Biol Interact 164:157–166. doi:10.1016/j.cbi.2006.09.007
Siemann EH, Creasy LL (1992) Concentration of the phytoalexin resveratrol in wine. Am J Enol Vitic 43:49–52
Frankel EN, Waterhouse AL, Kinsella JE (1993) Inhibition of human LDL oxidation by resveratrol. Lancet 341:1103–1104. doi:10.1016/0140-6736(93)92472-6
Pace-Asciak CR, Hahn S, Diamandis EP, Soleas G, Goldberg DM (1995) The red wine phenolics trans-resveratrol and quercetin block human platelet aggregation and eicosanoid synthesis: implications for protection against coronary heart disease. Clin Chim Acta 235:207–219. doi:10.1016/0009-8981(95)06045-1
Brito P, Almeida LM, Dinis TC (2002) The interaction of resveratrol with ferrylmyoglobin and peroxynitrite; protection against LDL oxidation. Free Radic Res 36:621–631. doi:10.1080/10715760290029083
Liu Y, Liu G (2004) Isorhapontigenin and resveratrol suppress oxLDL-induced proliferation and activation of ERK1/2 mitogen-activated protein kinases of bovine aortic smooth muscle cells. Biochem Pharmacol 67:777–785. doi:10.1016/j.bcp.2003.09.025
Dinis TC, Santos CL, Almeida LM (2002) The apoprotein is the preferential target for peroxynitrite-induced LDL damage protection by dietary phenolic acids. Free Radic Res 36:531–543. doi:10.1080/10715760290025915
Uppu RM, Pryor WA (1996) Biphasic synthesis of high concentrations of peroxynitrite using water-insoluble alkyl nitrite and hydrogen peroxide. Methods Enzymol 269:322–329. doi:10.1016/S0076-6879(96)69033-8
Denizot F, Lang R (1986) Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 89:271–277. doi:10.1016/0022-1759(86)90368-6
McConkey DJ, Hartzell P, Nicotera P, Orrenius S (1989) Calcium-activated DNA fragmentation kills immature thymocytes. FASEB J 3:1843–1849
Kapuscinski J, Skoczylas B (1977) Simple and rapid fluorimetric method for DNA microassay. Anal Biochem 83:252–257. doi:10.1016/0003-2697(77)90533-4
Scaduto RC Jr, Grotyohann LW (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 76:469–477
Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116:205–219. doi:10.1016/S0092-8674(04)00046-7
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407:770–776. doi:10.1038/35037710
Riedl SJ, Salvesen GS (2007) The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol 8:405–413. doi:10.1038/nrm2153
Kroemer G, Reed JC (2000) Mitochondrial control of cell death. Nat Med 6:513–519. doi:10.1038/74994
Sharpe JC, Arnoult D, Youle RJ (2004) Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta 1644:107–113. doi:10.1016/j.bbamcr.2003.10.016
Zhuang S, Simon G (2000) Peroxynitrite-induced apoptosis involves activation of multiple caspases in HL-60 cells. Am J Physiol Cell Physiol 279:C341–C351
Whiteman M, Armstrong JS, Cheung NS, Siau JL, Rose P, Schantz JT et al (2004) Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J 18:1395–1397
Szabo C, Ischiropoulos H, Radi R (2007) Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6:662–680. doi:10.1038/nrd2222
Quijano C, Alvarez B, Gatti RM, Augusto O, Radi R (1997) Pathways of peroxynitrite oxidation of thiol groups. Biochem J 322:167–173
Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A et al (1996) Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med 184:1155–1160. doi:10.1084/jem.184.3.1155
Petit PX, Goubern M, Diolez P, Susin SA, Zamzami N, Kroemer G (1998) Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett 426:111–116. doi:10.1016/S0014-5793(98)00318-4
Virag L, Szabo C (2000) BCL-2 protects peroxynitrite-treated thymocytes from poly(ADP-ribose) synthase (PARS)-independent apoptotic but not from PARS-mediated necrotic cell death. Free Radic Biol Med 29:704–713. doi:10.1016/S0891-5849(00)00359-2
Choi JW, Yoo BK, Ryu MK, Choi MS, Park GH, Ko KH (2005) Adenosine and purine nucleosides prevent the disruption of mitochondrial transmembrane potential by peroxynitrite in rat primary astrocytes. Arch Pharm Res 28:810–815
Li MH, Cha YN, Surh YJ (2006) Carbon monoxide protects PC12 cells from peroxynitrite-induced apoptotic death by preventing the depolarization of mitochondrial transmembrane potential. Biochem Biophys Res Commun 342:984–990. doi:10.1016/j.bbrc.2006.02.046
Shacka JJ, Sahawneh MA, Gonzalez JD, Ye YZ, D’Alessandro TL, Estevez AG (2006) Two distinct signaling pathways regulate peroxynitrite-induced apoptosis in PC12 cells. Cell Death Differ 13:1506–1514. doi:10.1038/sj.cdd.4401831
Vieira HL, Belzacq AS, Haouzi D, et al (2001) The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene 20:4305–4316. doi:10.1038/sj.onc.1204575
Cassina A, Radi R (1996) Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys 328:309–316. doi:10.1006/abbi.1996.0178
Radi R, Cassina A, Hodara R, Quijano C, Castro L (2002) Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med 33:1451–1464. doi:10.1016/S0891-5849(02)01111-5
Gogvadze V, Orrenius S (2006) Mitochondrial regulation of apoptotic cell death. Chem Biol Interact 163:4–14. doi:10.1016/j.cbi.2006.04.010
Zhu L, Gunn C, Beckman JS (1992) Bactericidal activity of peroxynitrite. Arch Biochem Biophys 298:452–457. doi:10.1016/0003-9861(92)90434-X
Alvarez MN, Piacenza L, Irigoin F, Peluffo G, Radi R (2004) Macrophage-derived peroxynitrite diffusion and toxicity to Trypanosoma cruzi. Arch Biochem Biophys 432:222–232. doi:10.1016/j.abb.2004.09.015
Cao Z, Li Y (2004) Potent induction of cellular antioxidants and phase 2 enzymes by resveratrol in cardiomyocytes: protection against oxidative and electrophilic injury. Eur J Pharmacol 489:39–48. doi:10.1016/j.ejphar.2004.02.031
Das S, Cordis GA, Maulik N, Das DK (2005) Pharmacological preconditioning with resveratrol: role of CREB-dependent Bcl-2 signaling via adenosine A3 receptor activation. Am J Physiol Heart Circ Physiol 288:H328–H335
Pozo-Guisado E, Lorenzo-Benayas MJ, Fernandez-Salguero PM (2004) Resveratrol modulates the phosphoinositide 3-kinase pathway through an estrogen receptor alpha-dependent mechanism: relevance in cell proliferation. Int J Cancer 109:167–173. doi:10.1002/ijc.11720
Jang JH, Surh YJ (2003) Protective effect of resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free Radic Biol Med 34:1100–1110. doi:10.1016/S0891-5849(03)00062-5
Ahmad KA, Clement MV, Hanif IM, Pervaiz S (2004) Resveratrol inhibits drug-induced apoptosis in human leukemia cells by creating an intracellular milieu nonpermissive for death execution. Cancer Res 64:1452–1459. doi:10.1158/0008-5472.CAN-03-2414
Ou HC, Chou FP, Sheen HM, Lin TM, Yang CH, Huey-Herng SW (2006) Resveratrol, a polyphenolic compound in red wine, protects against oxidized LDL-induced cytotoxicity in endothelial cells. Clin Chim Acta 364:196–204. doi:10.1016/j.cccn.2005.06.018
Ungvari Z, Orosz Z, Rivera A et al (2007) Resveratrol increases vascular oxidative stress resistance. Am J Physiol Heart Circ Physiol 292:H2417–H2424. doi:10.1152/ajpheart.01258.2006
Clement MV, Hirpara JL, Chawdhury SH, Pervaiz S (1998) Chemopreventive agent resveratrol, a natural product derived from grapes, triggers CD95 signaling-dependent apoptosis in human tumor cells. Blood 92:996–1002
Babich H, Reisbaum AG, Zuckerbraun HL (2000) In vitro response of human gingival epithelial S-G cells to resveratrol. Toxicol Lett 114:143–153. doi:10.1016/S0378-4274(99)00288-X
Fontecave M, Lepoivre M, Elleingand E, Gerez C, Guittet O (1998) Resveratrol, a remarkable inhibitor of ribonucleotide reductase. FEBS Lett 421:277–279. doi:10.1016/S0014-5793(97)01572-X
Sun NJ, Woo SH, Cassady JM, Snapka RM (1998) DNA polymerase and topoisomerase II inhibitors from Psoralea corylifolia. J Nat Prod 61:362–366. doi:10.1021/np970488q
Acknowledgments
This work was supported by FCT (POCI/AGR/59919/2004). Paula Brito is a recipient of the grant SFRH/BD/7986/2001.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brito, P.M., Simões, N.F., Almeida, L.M. et al. Resveratrol disrupts peroxynitrite-triggered mitochondrial apoptotic pathway: a role for Bcl-2. Apoptosis 13, 1043–1053 (2008). https://doi.org/10.1007/s10495-008-0235-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0235-4