
Abstract
The retinal pigment epithelium (RPE) is constantly exposed to external injuries which lead to degeneration, dysfunction or loss of RPE cells. The balance between RPE cells death and proliferation may be responsible for several diseases of the underlying retina, including age-related macular degeneration (AMD) and proliferative vitreoretinopathy (PVR). Signaling pathways able to control cells proliferation or death usually involve the MAPK (mitogen-activated protein kinases) pathways, which modulate the activity of transcription factors by phosphorylation. UV exposure induces DNA breakdown and causes cellular damage through the production of reactive oxygen species (ROS) leading to programmed cell death. In this study, human retinal pigment epithelial cells ARPE19 were exposed to 100 J/m2 of UV-C and MAPK pathways were studied. We first showed the expression of the three major MAPK pathways. Then we showed that activator protein-1 (AP-1) was activated through phosphorylation of cJun and cFos, induced by JNK and p38, respectively. Specific inhibitors of both kinases decreased their respective activities and phosphorylation of their nuclear targets (cJun and cFos) and reduced UV-induced cell death. The use of specific kinases inhibitors may provide excellent tools to prevent RPE apoptosis specifically in RPE diseases involving ROS and other stress-related compounds such as in AMD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
UV radiation is part of the sunlight spectrum and is divided into three wave bands (UV-A, UV-B and UV-C). Experimental studies have used all three ranges of UV in various combinations and with multiple doses (see review [1]). The role of UV has been extensively studied in skin carcinogenesis because of the exposure of this organ. The eye is another directly exposed major organ and in contrary to the skin very little is known about the role of UV exposure on the retina. A recent study described the in vitro effect of UV-C irradiation on lens α-crystallin, a protein thought to play a role in maintaining lens transparency [2], whereas another study analyzed signaling pathways associated with corneal epithelial cell apoptosis [3]. All UV-radiations are genotoxic and even if human lenses differ from rat or mouse, components of UV are capable of reaching the retina, as shown by a structural study of the rat retina exposed to UV, where UB-B and UV-C produced alterations, which affected the function of the retina [4].
The MAPK (mitogen-activated protein kinases) constitute a major element in cellular signaling. Their distinctive feature is their ability to directly phosphorylate, and thereby modulate the activity of transcription factors that are targets of the initial stimuli [5]. In mammals, three major MAPK have been described: (1) the ERK1/2/3 (extracellular regulated kinases 1–3) [6], (2) the p38 kinases (p38 α/β/δ, and p38γ) also named ERK6 [7] and (3) the JNK1/2/3 (c-Jun N-terminal Kinases 1–3) [8]. Whereas ERKs are preferentially activated by growth factors and mitogens, JNKs and p38 kinases primarily respond to a variety of stimuli collectively designated as “stress-signals”. These stresses include UV exposure, osmotic shocks, oxidative stress and cytokine treatments (IL-1β, TNFα). Activation of the p38 and JNK cascades is associated with major changes in cell fate including growth arrest, apoptosis and activation of immune cells and it is believed that such activation dictates a functionally important fraction of the response in inflammatory and autoimmune diseases. MAPK signaling cascades are targets for UV and are important in the regulation of the multitude of UV-induced cellular responses (see review [1]).
Activated protein-1 (AP-1) transcription factor consists of a variety of dimers composed of the Fos (c-Fos, FosB, Fra1, Fra2), Jun (cJun, JunD, JunB) and CREB/ATF (ATF1, 2, 3, 4 and 5) families of proteins. AP-1 has been implicated in a large number of biological processes including cell differentiation, proliferation, apoptosis and oncogenic transformation. These families of proteins are downstream target of MAPK pathways and have been largely described to play a role in programmed cell death, especially in light-induced photoreceptor cell death [9]. Binding of AP-1 complex leads to activation or repression depending on the target gene and cellular context. These immediate early genes regulated by AP-1 are key players in the apoptotic mechanism of cell death.
Retinal pigmented epithelial (RPE) cells are highly polarized cells. Their integrity is critical for the maintenance of neural retina functions. In healthy subjects, RPE cells have limited potential of proliferation associated with growth and age, while in disease uncontrolled RPE cells proliferation may contribute to retinal diseases such as proliferative vitreoretinopathy (PVR). On the other hand, RPE is thought to be the prime early target for age-related macular degeneration (AMD), which involves RPE cells death and atrophy of the photoreceptors. Majors studies performed in RPE fields used a spontaneously arising human RPE cell line, ARPE19, as cellular model. ARPE19 cells exhibit a polarization and are capable of tight junction formation [10]. Previous studies have shown that overexpression of ERK1/2 confers resistance against RPE cells death mediated by oxidative stress [11], whereas inhibition of MEK1/2 completely abolished ERK1/2 activation and reduced cell proliferation [12]. Overactivation of the two other MAPK, p38 and JNK, was involved in RPE cell death induced by FCS-depleted cultures [13] and hydrogen peroxide exposure [14].
In this study, we showed for the first time in the human RPE cell line ARPE19, a link between UV-induced JNK and p38 activation, transactivation of AP-1 complex and ARPE19 cell apoptosis. By using specific kinase inhibitors, we were able to decrease kinase activation, AP-1 transactivation and consequently block UV-induced cell apoptosis.
Materials and methods
Animals
These studies adhered to the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of animals in ophthalmic and vision research and were approved by the Veterinary Service of the State of Valais (Switzerland). NMRI and C57BL/6 mice were purchased from Charles River (Les Oncins, France) and kept in a 12-h light-dark cycle with unlimited access to food and water.
Materials
Primary antibodies anti-phospho-JNK1/2 and anti-JNK1/2 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); anti-ERK2 were purchased from Abcam, Inc. (Cambridge, MA, USA); anti-phospho-ERK2, anti-phospho-p38, anti-p38, anti-phospho-cJun, anti-phospho-ATF2 were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA); and anti-tubulin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Secondary antibodies, anti-rabbit-HRP and anti-mouse-HRP were purchased from Amersham (United Kingdom). Alexa-Fluor 594 goat anti-rabbit antibody was purchased from Invitrogen AG (Basel, Switzerland). SB203580 inhibitor was purchased from Promega (Madison, USA). JNKi and Tat peptides were a gift from XigenPharma (Lausanne, Switzerland).
RPE cells isolation and cell culture
RPE cells were isolated from enucleated (C57BL/6 mice) eyes under microscope and lysed directly in loading buffer solution for western blot analysis. For in vitro studies, ARPE19 cells were cultured in DMEM-F12 medium (#31330, Invitrogen AG, Basel, Switzerland) complemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin [10]. The cells were grown at 37°C in a humidified 5% CO2 condition. Cultures were plates in 60 mm tissue culture-treated dishes to confluence. Passages 20–40 were used for this study. Two days after plating, the medium was changed to culture medium containing 10 μM SB203580, 10 μM Tat-D-JNKi or 10 μM Tat-peptide (control) as described before [15] for a 2-h period. Then ARPE19 cells were exposed to 100 J/m2, by using a UV CL-100 crosslinker (UVP, Inc. Cambridge, England), and incubated for diverse periods of time prior to be analyzed for protein expression or apoptosis. For experiment performed on whole eye, we enucleated the eye and exposed the entire organ to UV, then the eye was incubated in DMEM medium at 37°C for 30-min before RPE cells isolation. RPE cells were isolated under microscope; after retrieving the retina, melanin-containing cells were isolated mechanically and suspended in 50 μl sample buffer containing 20 mM Hepes, 0.5% Tween, phosphatase inhibitors cocktail 1 and 2 (Sigma #p2850, #p5726) and Complete protease inhibitors (Roche Applied Science, Rotkreus, Switzerland). Then cells were lysed with three successive freeze and thaw. Proteins content were quantified with Bradford assay before analysis.
Western blot analysis and AlphaScreen assay
ARPE19 cells were in culture medium condition as described above. At the end of the incubations, they were washed in PBS, suspended in 100 μl sample buffer containing 20 mM Hepes, 0.5% Tween, phosphatase inhibitors cocktail 1 and 2 (Sigma #p2850, #p5726) and Complete protease inhibitors (Roche Applied Science, Rotkreus, Switzerland). Then cells were lysed with three successive freeze and thaw. The detergent-insoluble material was pelleted by centrifugation at 15,000 rpm for 5 min at 4°C. The supernatants containing protein cell lysate were used for western blotting or AlphaScreen assays. For western blot analysis, proteins were electrically transferred to PVDF filters and incubated with anti-phospho-JNK1/2 (1/500), anti-JNK1/2 (1/2,000), anti-phospho-ERK2 (1/1,000), anti-ERK2 (1/5,000), anti-phospho-p38 (1/1,000), anti-p38 (1/400), anti-phospho-ATF2 (1/1,000), anti-phospho-cJun (1/1,000) and anti-tubulin (1/40,000) antibody as primary antibodies. Secondary antibodies, anti-rabbit-HRP (1/25,000) and anti-mouse-HRP (1/15,000) were used to detect protein expression. Immune complexes were detected by chemiluminescence using LumiGLO (Amersham). For AlphaScreen assay, P-p38 and P-ERK2 were quantified using SureFire kit (#TGR38S500 and #TGRES500, TGR BioSciences, Australia) as described by manufactors; briefly 5 μg of protein lysates were incubated during 1–2 min in activation buffer, then reaction buffer containing both anti-kinase and anti-phospho kinase antibodies were mixed with AlphaScreen donor and acceptor beads and added to activated protein lysates. After 2 h of incubation, AlphaScreen signal was measured on Envision® system. The measurement of JNK activity was performed as described previously [16]; briefly kinase reagents (10 nM B-GST-cJun, 30 nM anti P-cJun antibody and 50 μM of ATP) were first diluted in kinase buffer (20 mM Tris–HCl pH 7.6, 10 mM MgCl2, 1 mM DTT, 100 μM Na3VO4, 0.01% Tween-20) and added to 1 μg of proteins for 30 min at 23°C. Detection was performed by an addition of 10 μl of beads mix (Protein A acceptor 20 μg/ml and Streptavidin donor 20 μg/ml), diluted in detection buffer (20 mM Tris–HCl pH 7.4, 20 mM NaCl, 80 mM EDTA, 0.3% BSA), followed by a 1 h incubation at 23°C in the dark. The AlphaScreen signal was analyzed directly on EnVision® system.
AP-1 binding assay
ARPE19 cells were in culture medium condition as described above. At the end of the incubation period, nuclear extracts were prepared as previously described; briefly, cells were suspended in 1 ml cold buffer A (10 mM Hepes, pH 7.9, 10 mM KCl, 1 mM DTT and protease inhibitors) and placed for 15 min on ice. Then 62.5 μl of 10% NP-40 were added and supernatant discarded after centrifugation. Nuclei were then lysed in buffer B (20 mM Hepes, pH 7.9, 400 mM NaCl, 1 mM DTT and protease inhibitors) and quantified for protein content. Eight micrograms of protein lysates were used for AP-1 binding assay with TransAM AP-1 kit (Active Motif, Carlsbad, CA, USA) as described by the manufacturer.
Immunostaining
Enucleated eyes (NMRI mice) were rapidly fixed in paraformaldehyde 4% during 45 min prior to overnight incubation in 0.6 M sucrose. Then eyes were mounted in Yazula and sections of 8 μm performed. Sections were stained for MAPKs protein expression with rabbit anti-p38 antibodies (dilution 1/2,000), anti-ERK2 (dilution 1/2,000) and JNK1/2 (dilution 1/1,000). FITC Alexa-Fluor 594 goat anti-rabbit antibody (dilution 1/2,000) was used as secondary antibody.
Cell death analysis
In situ cell death detection was performed 24 h after UV treatment by TUNEL technology as described by the manufacturer (#11684795910 Roche Applied Science, Rotkreus, Switzerland). Quantification of ARPE19 cells apoptosis was performed after various periods of time (12, 24 and 48 h) by staining with propidium iodide and Hoechst 33342. Cells were defined as apoptotic when they exhibited a condensed nuclear chromatin or a fragmented nuclear membrane when visualized with Hoechst 33342. Necrotic cells were characterized by nuclear PI staining but without condensed chromatin or fragmented nuclear membrane. Cells without apoptotic or necrotic features where considered viable. A minimum of 1,000 cells were counted in five different fields in each experiment and results were given in percentage of apoptotic cells versus viable cells.
Statistical analysis
All results were expressed as means ± SEM of the indicated number of experiments. Statistical significance was calculated with the Student’s t test.
Results
UV-C induced apoptosis of cultured ARPE19 cells
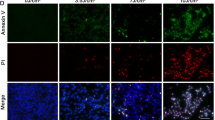
To study UV impact on ARPE19 cells, we cultured these cells in DMEM-F12 medium prior treatment by UV-C (100 J/cm2) during 10 s. Then cells were further cultured for diverse periods of time at 37°C and stained with Hoechst 33342 and PI for apoptosis analysis. We observed a time dependent apoptosis of ARPE19 cells induced by UV (Fig. 1a). Significant difference was observed 24 h after treatment with 3.69 ± 0.68% of apoptotic cells compared to 0.93 ± 0.33% for control. Apoptosis reached 8.8 ± 0.81% (0.77 ± 0.1% for control) of apoptotic cells 48 h after UV treatment. Panel B of Fig. 1 shows the increase of TUNEL positive cells 48 h after UV treatment. Arrow heads showed nuclear condensation at higher magnification which correspond to TUNEL positive cells.

UV-C induced apoptosis of cultured ARPE19 Cells. ARPE19 cells were cultured as described in Section ‘Materials and methods’. Then cells were treated by UV at 100 J/m2 and we measured apoptotic cells by TUNEL assay and by Hoechst and PI staining after UV treatment. (a) Percentage of ARPE19 cells presenting a condensed nuclear chromatin or a fragmented nuclear membrane were quantified by Hoechst and PI staining. Thousands of cells were counted in five different fields. The results were expressed as means ± SEM of three experiments. *p < 0.001 and **p < 0.002 versus CTL at the same period of time. (b) TUNEL assay experiment was performed to visualize ARPE19 apoptosis 48 h after UV treatment. White arrows indicate TUNEL positive cells and arrows head show a condensed nuclei (TUNEL positive) at higher magnification. This TUNEL assay is representative of three different experiments
The three major MAPKs are expressed in mouse RPE cells
To test the expression of JNK1/2, p38 and ERK2 MAPKs in RPE cells, we performed western blot analysis of whole mouse retina, isolated mouse RPE and human ARPE19 cells. As shown in Fig. 2, the three major MAPKs were expressed. It is interesting to notice that p38 was principally expressed in RPE cells as shown by western blot analysis (Fig. 2a) and immunostaining (Fig. 2b), whereas ERK2 was expressed in RPE cells, in outer nuclear layer (ONL), inner nuclear layer (INL) and ganglion cell layer (GCL) and JNK1/2 in RPE, INL and GCL.

The three major members of the MAPK family are expressed in RPE cells. (a) Expression of ERK2, JNK1/2, total p38 and tubulin was analyzed in isolated neuronal mouse retina, human ARPE19 cells and isolated mouse RPE cells by western blot analysis. (b) Immunostaining of the three kinases in NMRI mouse retina. Arrows showed the RPE cells of the retina. ONL: outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer
JNK1/2 and p38 are activated by UV treatment
To determine the role of MAPKs in UV-induced RPE cells apoptosis, we tested the phosphorylation state of those kinases, 30 min after UV treatment. As shown by western blot analysis (Fig. 3a), no phosphorylation of either JNK1/2 or total p38 was observed in control cells, but UV exposure clearly induced phosphorylation of both JNK1/2 and p38 kinases. ERK2 was highly phosphorylated at basal level (control condition) and slightly decreased by UV. The expression of non-phosphorylated kinases was not changed after UV stress for all three tested kinases. To confirm western blot observations, we measured the activity of JNKs by a non-radioactive AlphaScreen assay [16]. In this assay, the luminescence of the acceptor beads was proportional to JNKs activity, as shown in panel B of Fig. 3, where JNK activity was largely induced by UV. Because a similar kinase assay was not available to measure p38 and ERK2 activity, we used this AlphaScreen techniques to measure endogenous phosphorylation of both kinases (Fig. 3c). In this assay, the transfer of luminescence from donor to acceptor beads is proportional to the phosphorylation state of the kinase. Both assays correlated with the western blot analysis, and showed a high increase of p38 phosphorylation (700% increase vs. control; p < 0.002) and a significant decrease of ERK2 phosphorylation (30% decrease vs. control; p < 0.002) after UV treatment (Fig. 3c). Time course analysis was performed on ARPE19 cells and showed a rapid increase of JNK1/2 phosphorylation after 5 min with a sustained effect until 30 min and a decrease until 2 h, whereas the higher level of p38 phosphorylation occurred at 30 min and was maintained until 2 h (Fig. 4).

JNK1/2 and total p38 are phosphorylated after UV treatment. ARPE19 cells were cultured as described in Section ‘Materials and methods’. Then cells were treated 10 s by UV (100 J/m2) and protein extraction were performed 30 min after UV treatment. Activation of kinases was analyzed by western blot analysis (a) and quantification was performed by JNK AlphaScreen assay (b) and by AlphaScreen SureFire kit which measured endogenous P-ERK2 and total P-p38 (c) as described in Section ‘Materials and methods’. Western blot are representative of three distinct experiments; same samples were analyzed by AlphaScreen assays. The results are expressed as means ± SEM of three experiments. *p < 0.002 UV versus CTL. (d) Enucleated whole eye are treated by UV, as described for ARPE19 cells (100 J/m2) and cultured for 30 min at 37°C prior RPE cells isolation and protein expression analysis. RPE cells were isolated form the whole retina and western blot analysis was performed to analyze the phosphorylation state of JNK. These results are representative of two distinct experiments

Time curse of UV-induced JNK and p38 phosphorylation. ARPE19 cells were cultured as described in Section ‘Materials and methods’. Cells were treated by UV (100 J/m2) and protein extraction were performed at different time. Phosphorylation of JNK and p38, as well as tubulin expression was assess by western blot analysis. These results are representative of two distinct experiments
JNK2 is induced by UV in RPE mouse retina
To validate the use of the ARPE19 cells as a working model, we tested the effect of UV treatment on the whole mouse eye. Eyes were enucleated and treated by UV for 10 s, then the whole eye was incubated for 30 min in DMEM-F12 medium at 37°C. RPE cells were isolated and the phosphorylation state of JNK1/2 was tested by western blot analysis. Figure 3d shows the increase of JNK1/2 expression after UV exposure whereas tubulin expression was not changed. Interestingly, only JNK2 isoform phosphorylation, not JNK1, was increased by UV stress.
Inhibition of both JNK and p38 pathways prevented UV-induced RPE cell apoptosis
To determine a direct implication of JNK pathways in the UV-induced ARPE19 cells apoptosis we used a specific JNK inhibitor peptide (D-JNKi) which entered the cell and blocked JNK activation and cJun phosphorylation [15]. As shown in Fig. 5a and quantified in Fig. 5b, phosphorylation of JNK1/2 after UV treatment was drastically decreased when 10 μM of JNKi peptide was added 2 h prior the UV stress. Transfection of the control peptide (Tat) did not affect the expression or the phosphorylation state of JNK1/2. A slight but non-significant decrease of JNK1/2 expression was observed after JNK inhibition (Fig. 5).

Specific kinase inhibitors decreased JNK and p38 activity. ARPE19 cells were cultured as described in Section ‘Materials and methods’. Then proteins were extracted 30 min after UV treatment and western blot analysis (a, c) were performed to analyze kinase phosphorylation. (b) Quantification of the ratio phospho-kinase/kinase was performed on western blot by densitometry analysis. The results are expressed as means ± SEM of three experiments. *p < 0.005 CTL versus UV; **p < 0.02 UV/Tat versus UV/JNKi. (d) Measurement of endogenous phospho-p38 by AlphaScreen SureFire as described in Section ‘Materials and methods’. The results are expressed as means ± SEM of three experiments. *p < 0.0001 CTL versus UV; **p < 0.0001 UV versus UV/SB
As both JNK and p38 pathways were activated by UV, we tested the implication of p38 in this apoptotic mechanism. Using SB203580, an inhibitor of p38α/β, we first showed that UV-induced phosphorylation of p38 was decreased after inhibition (Fig. 5c). Then, by Alphascreen assay, we confirmed and quantified the induction of p38 phosphorylation by UV (Fig. 5d; six-fold induction by UV, p < 0.0001), and the inhibitory effect of SB203580, which decreased the phosphorylation of p38 (Fig. 5d; 60% of reduction compared with UV, p < 0.0001).
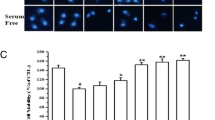
More interestingly, the decreased of JNK1/2 and p38 phosphorylation was accompanied by a strong reduction of UV-induced ARPE19 cells apoptosis, as measured and quantified by Hoechst/PI staining (Fig. 6a) and seen by TUNEL assays (Fig. 6b). Apoptosis reached 10.21 ± 1.1% (1.04 ± 0.07% for control) of apoptotic cells 48 h after UV treatment; JNK inhibition reduced cell death to 2.94 ± 0.17%, while p38 inhibition reduced cell death to 3.9 ± 0.4%. Similar results were obtained by quantification (data not shown) of TUNEL positive cells shown in Fig. 6b. Both, JNK and p38, inhibitors decreased level of UV-induced apoptosis, which clearly supports a role of both kinases in this apoptotic mechanism.

Specific JNK and p38 kinase inhibitors decreased UV-induced apoptosis. ARPE19 cells were cultured as described in Section ‘Materials and methods’. (a) Percentage of ARPE19 cells presenting a condensed nuclear chromatin or a fragmented nuclear membrane were quantified by Hoechst and PI staining. Thousands of cells were counted in five different fields. The results were expressed as means ± SEM of three experiments. **p < 0.0001 versus CTL and *p < 0.0006 versus UV. (b) Apoptosis analysis by TUNEL assay was performed 48 h after UV treatment according to guideline protocol. White arrows indicate TUNEL positive cells. Experiments were representative of three distinct experiments
This mechanism may involve AP-1 complex
The activator protein-1 (AP-1) transcription factor is composed of a mixture of heterodimeric complexes of proteins derived from the Fos, Jun and ATF families. The role of cFos and cJun, two partners of the AP-1 complex and known targets of p38 and JNK, were evaluated in this UV model. As shown by western blot analysis (Fig. 7a) the phosphorylation of cJun was induced by JNK activation, 30 min after UV treatment, whereas inhibition of JNK decreased this phosphorylation. No effect was observed in presence of the Tat peptide control. Since no high-quality anti-phospho cFos antibody was available, we tested the activation of cFos with an AP-1 binding assay. Figure 7b shows the increase of cFos binding to AP-1 sequence 6 h after UV stress. Inhibition of p38 with SB203850 significantly decreased the AP-1 binding of cFos. AP-1 binding of the other members of Jun and Fos families were not changed after UV stress (data not shown). To exclude a role of ATF2 in the AP-1 complex, we tested the effect of both JNK and p38 inhibitors in the UV-induced ATF2 phosphorylation. Figure 7c clearly shows the induction of ATF2 phosphorylation by UV, while a small but not significant effect of both inhibitors was observed.

UV-induced cJun and cFos phosphorylation activated AP-1. ARPE19 cells were cultured as described in Section ‘Materials and methods’. ARPE19 cell were then treated with UV and incubated for 30 min at 37°C prior western blot analysis for phospho-cJun (a) and phospho-ATF2 (c) or for 6 h for cFos AP-1 binding analysis (b). Eight micrograms of nuclear extract proteins were used for AP-1 binding assay as described in Section ‘Materials and methods’. The results are expressed as means ± SEM of three experiments. *p < 0.0001 CTL versus UV/Tat or UV; **p < 0.0001 UV versus UV/JNKi and # p < 0.04 UV versus UV/SB
Discussion
Retinal pigment epithelium (RPE) cells play a critical role in the neural retina by maintaining functional and healthy photoreceptors. These cells may experience various extracellular stimuli which promote survival or cell death through highly regulated signaling. UV exposure is one of this external stress agent and UV-induced MAPKs activity has largely been documented in human prostate adenocarcinoma DU-145 cells [17], human keratinocytes [18, 19], human embryonic kidney 293 cells [20], mouse epidermal JB6 cells [21], mouse embryonic fibroblasts [22] and corneal epithelial cells (see review [1] and [3, 23]). Surprisingly, few studies were performed on retina or RPE cells. In this study we focused on the effect of UV on RPE cells and deciphered the mechanism of UV-induced RPE cells apoptosis. Human RPE cell line (ARPE19) is not similar to primary cultured RPE cells. Despite these differences, this cell line has extensively been studied to obtain important insights into RPE cell biology [14]. Recently, Cai et al. compared gene expression profile in adult isolated human RPE and ARPE19 cells line and showed no clear difference in the global expression of genes known to be involved in angiogenesis, phagocytosis and apoptosis [24]. This and other studies supported the use of human ARPE19 cells as a model to analyze mechanism of RPE cells death. The UV-activated signal transduction pathways are mediated primarily through signaling cascades involving MAPKs, including ERK, JNK and p38 kinases which control the activities of various transcription factors [1]. Herein, we confirmed in ARPE19 cells, the preponderate role of either JNK or p38 in the transactivation of AP-1 transcription factor, via the phosphorylation of cJun and cFos. The activation of both pathways triggered the UV-induced apoptotic process of ARPE19 cells. Specific inhibitors of JNK and p38 were sufficient to block 70–60% of UV-induced apoptosis. Further studies using isolated human RPE cells instead of human ARPE19 cell line will be needed to confirm the role of AP-1 in UV-induced RPE cell death.
Several studies showed an activation of ERK1/2 pathway by UV [11, 21, 25, 26]. In contrast, we observed a slight decrease of phospho-ERK1/2 in ARPE19 cells. This decrease fit well with the low proliferation rate of cells observed after the UV stress (data not shown) and with the study of Hecquet et al. where inhibition of ERK1/2 by cAMP inhibited RPE cells proliferation [12]. In our cellular model, ERK1/2 may have played an anti-apoptotic role and therefore the observed decrease could have amplified the apoptotic effect of the two other kinases, JNK and p38.
As it is generally accepted that ERK1/2 activation is essential for cell survival, whereas activation of JNK and p38 is thought to play a role in death signaling [27]; therefore we focused on the JNK and p38 pathways. Since fibroblasts from JNK1−/− JNK2−/− double knock-out mice are resistant to UV-induced cell death [28], we postulated that both JNK isoforms were essential for apoptosis. Indeed, we observed activation of JNK1/2 after UV exposure as reported in many other cell systems [3, 14, 17, 19, 21, 27, 29–32]. On the other hand, only phosphorylation of JNK2, not JNK1, was observed in whole eye exposed to UV. These results suggest that JNK2 is the major and primary isoform responsible for the phosphorylation/activation of cJun in mouse RPE. However, in its absence, JNK1 could substitute for and phosphorylate cJun [33]. Further analyses, also including the neuronal JNK3, will be needed to dissect the various contributions of each of these isoforms in UV-induced apoptosis.
p38 is usually associated with JNK and has been implicated in numerous stress-induced apoptosis [1, 13, 20, 31, 34]. In this study, we found an activation of this pathway after UV stress and specific inhibition of p38 blocked the UV-induced apoptosis. There are four different isoforms (α, β, δ and γ) of p38 kinase, but a specific expression profile of each isoform in the retina as well as in the RPE cells line has not been performed, yet. Therefore we measured the activation of total p38 either by western blot or by AlphaScreen techniques. As SB203580 has been shown to efficiently inhibit the α and β, but much less the γ and δ isoforms [35], the α and β isoforms may be involved in UV-induced ARPE19 cells apoptosis. Development of isoform specific kinase assays is needed to precisely determine which p38 isoform is implicated in this mechanism.
The transcription factor AP-1 consists of a variety of dimers composed of the Fos, Jun and CREB/ATF families of proteins, which are potential JNK and p38 targets. It has been implicated in a large number of biological processes including cell differentiation, proliferation and apoptosis [36]. Phosphorylation of cJun by JNK is required for UV effect, because mouse embryonic fibroblasts cells isolated from cJunAA63/73/cJunAA63/73 mouse, a mutant with non-phosphorylable c-Jun, were insensitive to UV-induced cell death [37]. Moreover inhibition of cJun activity suppressed apoptosis whereas overexpression of cJun leads to cells apoptosis [38]. Herein, we were able to show by western blot analysis a strong phosphorylation of cJun after UV treatment. The use of a very specific JNK inhibitor peptide [39] was able to decrease this phosphorylation and the related UV-induced apoptosis. These results strongly argue for a role of JNK-activated cJun in the UV-induced RPE cells death.
Specific inhibition of p38 was also accompanied by a major decrease of cells apoptosis, similar to that observed with JNK inhibition. Therefore we tested for ATF2 and cFos activation, two other AP-1 binding partners of cJun described as potential targets of p38 kinase. ATF2 has been described to be phosphorylated either by ERK2, JNK or p38 [40]. In our cell model, UV induced ATF2 phosphorylation, but inhibitors of JNK and p38 failed to significantly block this activation. We could suspect that the high level of phospho-ERK2, present in the cell after UV stress, could be sufficient to activate ATF2. Since ATF2 phosphorylation was still high, despite a strong reduction in UV-induced apoptosis following inhibition of JNK and p38, we ruled out this nuclear factor as a main actor of this mechanism. Interestingly, we detected a reactive band induced by UV (lower band ∼45 kDa Fig. 7c) with the anti phospho-ATF2 antibody, which could be a specific ATF2 isoform. Further analysis will be needed to investigate this result.
We then looked for the activation of Fos family members, other potential partners of cJun nuclear factor. Recent studies showing a direct activation of cFos by p38 in HEK 293T cells after a UV stress [20] and the fact that absence of cFos prevents light induced apoptosis [9], suggest that cFos could be involved in UV-induced apoptosis. We were able to show an activation of cFos binding to AP-1 sequence, 6 h after UV stress. AP-1 binding of the other members of Fos family (FosB, Fra1 and 2, FosD) was not changed (data not shown). The inhibition of cFos binding to AP-1 with SB203850 was partial but very similar to previously reported data on HEK-293T cells treated by UV [22]. This result, together with the incomplete inhibition of phospho-cJun by JNKi suggest that UV-induced apoptosis is an AP-1 dose, possibly threshold dependent mechanism. An approach using siRNA to decrease either cJun or cFos, or both nuclear factors expression may be used to confirm the specific role of AP-1 in such mechanism.
Although we described the mechanisms by which UV induced ARPE19 cells apoptosis, we still do not know how UV light is sensed by the cells and which final targets of AP-1 are involved in the cell death program. One hypothesis, besides DNA breakage, is that UV acts directly by producing of reactive oxygen species (ROS) [41], which leads to the activation of MAPK signaling pathways [42]. Retina is an ideal environment for generation of ROS: oxygen consumption is high, photoreceptors are composed of polyunsaturated fatty acids readily oxidized, RPE cells contain an abundance of photosensitizers and finally the photoreceptors phagocytosis process generates high level of ROS by itself. Many MAPK pathways including the apoptosis signal-regulating kinase 1 (ASK1) are activated by ROS and a body of evidence suggests that ASK-1 could be the major actor of this apoptotic process. ASK1 is able to activate both JNK and p38 pathways [43, 44] and deletion of ASK1 in mice protects against TNF- and H2O2-induced apoptosis in fibroblast and prevents prolonged activation of JNK and p38 [42]. The use of specific inhibitors of ASK-1 will provide more insight into its role in this apoptotic process. Finding downstream targets of AP-1 transactivator will also be a challenge to fully describe this mechanism. The family of Foxo genes could be a target, as they have been described to be potential effectors of p38. Moreover FOXO1 mediates transcriptional induction of the pro-apoptotic molecule, Bim [45] and has been implicated in UV-induced DNA damage in the developing Drosophila retina [32]. JNK phosphorylates also non-transcription factors such as members of the Bcl-2 family (Bcl-2, Bcl-XL, Bax, Bim and Bad) which have been implicated in apoptotic processes [46]. Furthermore, both JNK and p38 are able to phosphorylate BAX, consequently promoting its mitochondrial translocation and triggering apoptosis [47].
Conclusion
The role of oxidative stress (call the “free radical theory” in the middle of the twentieth century [48]) has been implicated in many age-related diseases, where it was generally induced by multiple environmental stimuli including cytokines, UV radiation, chemotherapeutic agents and other stresses. Large numbers of studies support a key role of reactive oxygen species in AMD (see review [49]). Deciphering the whole mechanism of RPE cells apoptosis with a special focus on this mechanism in AMD pathology will give us important insight to understand processes involved in aging pathologies. It will be interesting to test MAPKs activity (JNK, p38 and ASK1) and ROS production in RPE cells of AMD mouse models described by Ambati et al. [50] and Tuo et al. [49], as well as directly on retina from AMD patients. Modulation of RPE cell proliferation or death with the use of specific inhibitors or selective activators may be useful to reduce oxidative damage occurring in age-related diseases.
References
Bode AM, Dong Z (2003) Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci STKE 2003:RE2
Fujii N, Uchida H, Saito T (2004) The damaging effect of UV-C irradiation on lens alpha-crystallin. Mol Vis 10:814–820
Lu L, Wang L, Shell B (2003) UV-induced signaling pathways associated with corneal epithelial cell apoptosis. Invest Ophthalmol Vis Sci 44:5102–5109
Campos de Oliviera Miguel N, Meyer-Rochow VB, Allodi S (2003) A structural study of the retinal photoreceptor, plexiform and ganglion cell layers following exposure to UV-B and UV-C radiation in the albino rat. Micron 34:395–404
Widmann C, Gibson S, Jarpe MB, Johnson GL (1999) Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev 79:143–180
Davis RJ (1995) Transcriptional regulation by MAP kinases. Mol Reprod Dev 42:459–467
Enslen H, Brancho DM, Davis RJ (2000) Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J 19:1301–1311
Davis RJ (1994) MAPKs: new JNK expands the group. Trends Biochem Sci 19:470–473
Grimm C, Wenzel A, Hafezi F, Reme CE (2000) Gene expression in the mouse retina: the effect of damaging light. Mol Vis 6:252–260
Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM (1996) ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res 62:155–169
Glotin AL, Calipel A, Brossas JY, Faussat AM, Treton J, Mascarelli F (2006) Sustained versus transient ERK1/2 signaling underlies the anti- and proapoptotic effects of oxidative stress in human RPE cells. Invest Ophthalmol Vis Sci 47:4614–4623
Hecquet C, Lefevre G, Valtink M, Engelmann K, Mascarelli F (2002) cAMP inhibits the proliferation of retinal pigmented epithelial cells through the inhibition of ERK1/2 in a PKA-independent manner. Oncogene 21:6101–6112
Hecquet C, Lefevre G, Valtink M, Engelmann K, Mascarelli F (2003) Activation and role of MAP kinase-dependent pathways in retinal pigment epithelium cells: JNK1, P38 kinase, and cell death. Invest Ophthalmol Vis Sci 44:1320–1329
Ho TC, Yang YC, Cheng HC et al (2006) Activation of mitogen-activated protein kinases is essential for hydrogen peroxide-induced apoptosis in retinal pigment epithelial cells. Apoptosis 11:1899–1908
Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF (2001) Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50:77–82
Guenat S, Rouleau N, Bielmann C et al (2006) Homogeneous and nonradioactive high-throughput screening platform for the characterization of kinase inhibitors in cell lysates. J Biomol Screen 11:1015–1026
Song JJ, Lee YJ (2006) Differential activation of the JNK signal pathway by UV irradiation and glucose deprivation. Cell Signal 19:563–572
Chen W, Tang Q, Gonzales MS, Bowden GT (2001) Role of p38 MAP kinases and ERK in mediating ultraviolet-B induced cyclooxygenase-2 gene expression in human keratinocytes. Oncogene 20:3921–3926
Silvers AL, Bachelor MA, Bowden GT (2003) The role of JNK and p38 MAPK activities in UVA-induced signaling pathways leading to AP-1 activation and c-Fos expression. Neoplasia 5:319–329
Tanos T, Marinissen MJ, Leskow FC et al (2005) Phosphorylation of c-Fos by members of the p38 MAPK family. Role in the AP-1 response to UV light. J Biol Chem 280:18842–18852
Zhu F, Zhang Y, Bode AM, Dong Z (2004) Involvement of ERKs and mitogen- and stress-activated protein kinase in UVC-induced phosphorylation of ATF2 in JB6 cells. Carcinogenesis 25:1847–1852
Zhou H, Gao J, Lu ZY, Lu L, Dai W, Xu M (2007) Role of c-Fos/JunD in protecting stress-induced cell death. Cell Prolif 40:431–444
Shi B, Isseroff RR (2006) Arsenite pre-conditioning reduces UVB-induced apoptosis in corneal epithelial cells through the anti-apoptotic activity of 27 kDa heat shock protein (HSP27). J Cell Physiol 206:301–308
Cai H, Del Priore LV (2006) Gene expression profile of cultured adult compared to immortalized human RPE. Mol Vis 12:1–14
Kimoto K, Nakatsuka K, Matsuo N, Yoshioka H (2004) p38 MAPK mediates the expression of type I collagen induced by TGF-beta 2 in human retinal pigment epithelial cells ARPE-19. Invest Ophthalmol Vis Sci 45:2431–2437
Hinton DR, He S, Graf K et al (1998) Mitogen-activated protein kinase activation mediates PDGF-directed migration of RPE cells. Exp Cell Res 239:11–15
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270:1326–1331
Tournier C, Hess P, Yang DD et al (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288:870–874
Bulavin DV, Higashimoto Y, Popoff IJ et al (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411:102–107
Liu H, Deng X, Shyu YJ, Li JJ, Taparowsky EJ, Hu CD (2006) Mutual regulation of c-Jun and ATF2 by transcriptional activation and subcellular localization. EMBO J 25:1058–1069
Aggeli IK, Gaitanaki C, Beis I (2006) Involvement of JNKs and p38-MAPK/MSK1 pathways in H2O2-induced upregulation of heme oxygenase-1 mRNA in H9c2 cells. Cell Signal 18:1801–1812
Luo X, Puig O, Hyun J, Bohmann D, Jasper H (2006) Foxo and Fos regulate the decision between cell death and survival in response to UV irradiation. EMBO J 26:380–390
Hochedlinger K, Wagner EF, Sabapathy K (2002) Differential effects of JNK1 and JNK2 on signal specific induction of apoptosis. Oncogene 21:2441–2445
Hazzalin CA, Cuenda A, Cano E, Cohen P, Mahadevan LC (1997) Effects of the inhibition of p38/RK MAP kinase on induction of five fos and jun genes by diverse stimuli. Oncogene 15:2321–2331
Cuenda A, Cohen P, Buee-Scherrer V, Goedert M (1997) Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/p38). EMBO J 16:295–305
Jochum W, Passegue E, Wagner EF (2001) AP-1 in mouse development and tumorigenesis. Oncogene 20:2401–2412
Behrens A, Sibilia M, Wagner EF (1999) Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet 21:326–329
Ham J, Babij C, Whitfield J et al (1995) A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron 14:927–939
Borsello T, Clarke PG, Hirt L et al (2003) A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med 9:1180–1186
Kyriakis JM (2000) MAP kinases and the regulation of nuclear receptors. Sci STKE 2000:E1
Herrling T, Fuchs J, Rehberg J, Groth N (2003) UV-induced free radicals in the skin detected by ESR spectroscopy and imaging using nitroxides. Free Radic Biol Med 35:59–67
McCubrey JA, Lahair MM, Franklin RA (2006) Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal 8:1775–1789
Tobiume K, Matsuzawa A, Takahashi T et al (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2:222–228
Nagai H, Noguchi T, Takeda K, Ichijo H (2007) Pathophysiological roles of ASK1-MAP kinase signaling pathways. J Biochem Mol Biol 40:1–6
Greer EL, Brunet A (2005) FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24:7410–7425
Schinzel A, Kaufmann T, Borner C (2004) Bcl-2 family members: integrators of survival and death signals in physiology and pathology [corrected]. Biochim Biophys Acta 1644:95–105
Kim BJ, Ryu SW, Song BJ (2006) JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem 281:21256–21265
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247
Beatty S, Koh H, Phil M, Henson D, Boulton M (2000) The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol 45:115–134
Tuo J, Bojanowski CM, Zhou M et al (2007) Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci 48:3827–3836
Acknowledgments
We thank Aurélie Despont, Martine Emery and Loriane Moret for technical help. We thank Dr. Sandra Cottet and Dr. Pascal Escher for critical comments and helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Roduit, R., Schorderet, D.F. MAP kinase pathways in UV-induced apoptosis of retinal pigment epithelium ARPE19 cells. Apoptosis 13, 343–353 (2008). https://doi.org/10.1007/s10495-008-0179-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0179-8