
Abstract
The rumen microbiome contributes greatly to the degradation of plant fibres to volatile fatty acids and microbial products, affecting the health and productivity of ruminants. In this study, we investigated the dynamics of colonisation by bacterial communities attached to reeds and cottonseed hulls in the rumen of Tarim red deer, a native species distributed in the desert of the Tarim Basin. The reed and cottonseed hull samples incubated in nylon bags for 1, 6, 12, and 48 h were collected and used to examine the bacterial communities by next-generation sequencing of the bacterial 16S rRNA gene. Prevotella1 and Rikenellaceae RC9 were the most abundant taxa in both the reed and cottonseed hull groups at various times, indicating a key role of these organisms in rumen fermentation in Tarim red deer. The relative abundances of cellulolytic bacteria, such as members of Fibrobacter, Treponema 2, Ruminococcaceae NK4A214 and Succiniclasticum increased, while that of the genus Prevotella 1 decreased, with increasing incubation time in both reeds and cottonseed hulls. Moreover, the temporal changes in bacterial diversity between reeds and cottonseed hulls were different, as demonstrated by the variations in the taxa Ruminococcaceae UCG 010 and Papillibacter in the reed group and Sphaerochaeta and Erysipelotrichaceae UCG 004 in the cottonseed hull group; the abundances of these bacteria first decreased and then increased. In conclusion, our results reveal the dynamics of bacterial colonisation of reeds and cottonseed hulls in the rumen of Tarim red deer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ruminant livestock sector is an important part of the agricultural system because it provides meat and milk for human consumption; however, this sector is challenged by increasing demand from the growing human population (Eisler et al. 2014). Therefore, improvement of ruminant productivity is important for human food security. The rumen is the main location of degradation of the ingested diet, particularly forage, and is one of the most rapid and efficient plant fibre utilisation systems known (Russell and Rychlik 2001). The efficiency of the rumen depends on the diversity and density of microorganisms that live in this organ, including bacteria, protozoa, fungi and archaea (Russell and Rychlik 2001). Among these microorganisms, bacteria are the most abundant, greatly contributing to the digestion and conversion of feeds into volatile fatty acids and microbial products (Kim et al. 2011). On the other hand, the rumen microbiota is also considered a promising resource for the production of alternative biofuels, green chemicals and biomaterials due to the high conversion of lignocellulose in rumen (Hess et al. 2011; Svartström et al. 2017). Therefore, understanding the interaction between the microbiota and plant fibres in the rumen is important for improvement of ruminant productivity and biofuel production.
The rumen ecological system consists of three fractions, namely, a solid-adherent fraction, a liquid fraction and the rumen epithelium. Previous studies have demonstrated that the microbial community in the solid fraction is significantly different from that in the liquid fraction (De Mulder et al. 2017; Larue et al. 2005; McAllister et al. 1994), suggesting key roles in fibre degradation. However, the attachment of the microbiota to plant fibres has been shown to be a dynamic process (Huws et al. 2013, 2016; Jin et al. 2018; Piao et al. 2014), consisting of two stages. In the first stage, the primary colonising bacteria are likely to utilise soluble nutrients and, in the second stage, the colonisers efficiently degrade plant structural components (Huws et al. 2016). In addition, the rumen microbiota is affected by the ruminant hosts (Henderson et al. 2015) and the attached microbiota is also affected by the forage type (Ding et al. 2015; Liu et al. 2016). Therefore, examination of the temporal microbiota attached to plant fibres in different hosts could further improve our knowledge regarding microbiota-plant interactions.
Tarim red deer (Cervus elaphus yarkandensis), living in the deserts of the Tarim Basin, is the only subspecies of red deer in Central Asia that exhibits unique adaptation to a dry climate (Tumur et al. 2013). Tarim red deer are also listed as an endangered species by the International Union for the Conservation of Nature and Natural Resources (IUCN) due to the decreasing populations of this species (Brook et al. 2017). In nature, Tarim red deer are concentrated in reed (Phragmites communis) and poplar (Populus diversifolia) meadows (Qiao et al. 2006). However, in Tarim red deer husbandry, cottonseed hulls, a by-product of cotton processing that contains a large proportion of neutral detergent fibre (NDF) and associated lignin (Kononoff and Heinrichs 2003), are extensively used for velvet antler production. Thus, the rumen ecology of Tarim red deer may be a possible source for the discovery of efficient microbes that utilise plant fibres in harsh environments. In a previous study, we investigated the rumen bacteria of Tarim red deer (Qian et al. 2017); however, the mechanism via which the rumen microbiota colonises reeds and cottonseed hulls is largely unknown. Understanding the dynamics of the rumen microbiota in reeds and cottonseed hulls is important for improvement of the forage utilisation efficiency of Tarim red deer.
In this study, we tested the hypothesis that the rumen bacterial communities that colonise reeds and cottonseed hulls shift over time and identified the differences in the microbiota between reeds and cottonseed hulls.
Materials and methods
Animals and sample collection
Five healthy female rumen-cannulated Tarim red deer (average body weight = 150 ± 5.0 kg) were used in this study and maintained at the experimental animal centre of Tarim University. All the animals were assigned to individual metabolic pens and were fed twice each day, at 9:00 am and 7:00 pm, and had free access to clean drinking water. Each animal was fed a total of 4.2 kg of dry matter (75% cottonseed hulls, 25% corn-based diet) per day, with half of the allowed daily ration given at each feeding time. All animal care procedures were approved and authorised by the animal ethics committee of Tarim University.
The reeds and cottonseed hulls were dried at 65 °C and then milled through a 2.5 mm screen. The obtained forage samples were weighed into nylon bags with 38 µm pores (12 × 8 cm, 2.5 g/bag). Sixteen bags were placed in the rumen of each of the five red deer before the 9:00 am feeding (two bags for each forage at each time point). Two bags per red deer were recovered after 1, 6, 12, and 48 h, so a total of ten bags were obtained at each time point for each forage treatment. The bags were washed using sterile saline to remove the loosely attached microorganisms. The samples were preserved on dry ice and then transported to the laboratory for storage at − 80 °C until further analysis.
Chemical analysis
The dry matter (DM) and crude protein (CP) were determined according to AOAC methods (AOAC 2000). The NDF was determined using the procedures of van Soest et al. (1991).
DNA extraction, sequencing, data processing
Total genomic DNA was extracted from 200 mg of rumen-incubated reed and cottonseed hull samples using the QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) were used to amplify the bacterial 16S rRNA gene V4 region (Caporaso et al. 2012). The PCR was carried out using Phusion high-fidelity PCR master mix with GC buffer (New England Biolabs). The amplicons were quantified using a QuantiFluor®-P fluorometer (Promega, CA, USA) and sequenced on an Illumina MiSeq platform (Illumina Inc., San Diego, CA) to generate 250 bp paired-end reads.
The obtained raw sequences were first processed for quality control and filtering and then analysed using Quantitative Insights into Microbial Ecology (QIIME) ver. 1.9.1 (Caporaso et al. 2010b). Briefly, the chimeric sequences were detected and removed using Uchime software (Edgar et al. 2011). The remaining sequences were classified into operational taxonomic units (OTUs) based on a 97% similarity cutoff using UPARSE (Edgar 2013). The representative sequences of each OTU (the most abundant sequences within each OTU) were assigned by comparison against the SILVA database (version 128) using PyNAST (Caporaso et al. 2010a; Quast et al. 2013). After removing the singleton sequences, the data for reach sample were rarefied to the minimum value for all samples. A phylogenetic tree was constructed using FastTree (Price et al. 2009). The Good’s coverage, Shannon–Wiener, and Chao 1 indices for all the samples were also calculated using QIIME ver. 1.9.1 (Caporaso et al. 2010b).
Bioinformatics and statistical analyses
The bacterial community structures at all time points for each forage were compared using principal coordinate analysis (PCoA) based on the unweighted unifrac distances (which examines the presence and absence of bacterial lineages) and weighted unifrac distances (which accounts for the relative abundances of bacterial lineages) (Lozupone et al. 2011). Then, a canonical correlation analysis (CCA) was performed to identify the differences in the bacterial communities from reeds and cottonseed hulls at each time point using the RAM package (Dufrene and Legendre 1997). The relative abundances of these identified bacterial taxa were analysed using the general linear model (GLM) procedure of SPSS22 (SPSS, Chicago, IL, USA). If GLM tests indicated a significant difference between means, a one-way ANOVA was conducted to test the statistical significance of the taxonomic abundance based on the Benjamini–Hochberg corrected P value (false discovery rate < 0.05).
Accession numbers
The sequences from the present study have been deposited in the SRA database under accession number SRP116263.
Results
Biomass degradation of reed and cotton seed hulls after incubation in the rumen of Tarim red deer
The biomass degradation of reed and cottonseed hulls after incubation in the rumen of Tarim red deer were measured (Table 1). The results showed that both the DM, NDF and CP loss were significantly affected by incubation time (P < 0.0001). The DM of both reed and cottonseed hulls were degraded by approximately 6% after 1 h incubation. However, the difference between reed and cottonseed hulls was not significant over incubation time. The NDF disappearance was notably higher in cottonseed hulls than that in reed after 48 h incubation, while the CP loss was notably higher in reed than that in cottonseed hulls (Table 1).
Summary of 16S rRNA gene sequencing data
Overall, we obtained a total of 3,216,672 high-quality 16S rRNA gene sequences across all time points, with 1,482,328 and 1,734,344 sequences for the reed and cottonseed hull groups, respectively. Each sample had an average of 53,611 sequences. A total of 3403 and 3576 OTUs were identified with 97% sequence similarity in the reed and cottonseed hull groups, respectively (Fig. 1). For the reed group, the number of OTUs and the Chao1 and Shannon indices decreased from 1 to 12 h of incubation (Fig. 1A), although the differences were not significant. For the cottonseed hull group, the diversity indices increased from 1 to 12 h of incubation, and the differences between the values at 1 h and those at the other three time points were significant (Fig. 1B).

Comparison of the diversity of the bacteria attached to reeds (A) and cottonseed hulls (B) after incubation for 1, 6, 12 and 48 h in the rumen of Tarim red deer
Dynamics of the bacterial communities attached to reeds over time
The 3403 OTUs identified in the reed group were classified into a total of 19 phyla (Figure S1). The phyla Bacteroidetes, Firmicutes and Proteobacteria were the predominant bacteria regardless of incubation time, accounting for more than 80% of the detected OTUs. The abundance of the phylum Bacteroidetes (1 h = 74.2%, 6 h = 59.1%, 12 h = 57.3%, 48 h = 48.9%) decreased linearly with increasing incubation time, while that of the phylum Firmicutes increased (1 h = 11.9%, 6 h = 21.3%, 12 h = 16.4%, 48 h = 28.6%, respectively). Moreover, the relative abundance of the phylum Fibrobacteres increased linearly from 1 to 48 h of incubation (P < 0.05).
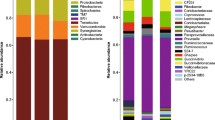
At the genus level, sequences were classified into 277 taxa across all rumen-incubated reed samples, and 49 taxa were identified with relative abundances greater than 0.5% at least one incubation time point, accounting for approximately 95% of the detected OTUs (Fig. 2A). After 1 h of incubation of reeds in the rumen of Tarim red deer, members of the genus Prevotella 1 (37.4%) were the most dominant bacteria, followed by bacteria belonging to Rikenellaceae RC9 (14.1%), Bacteroidales RF16 (7.2%), Succinivibrio (6.0%) and Prevotellaceae UCG003 (5.3%), accounting for approximately 65% of the overall bacterial composition. After incubation for 6 h, members of the genus Prevotella 1 (25.7%) remained the most dominant bacteria, followed by bacteria belonging to Rikenellaceae RC9 (15.4%), Prevotellaceae UCG001 (5.5%), and Anaeroplasma (4.0%), accounting for approximately 50% of the overall bacterial composition. After 12 h of incubation, members of the genus Prevotella 1 (20.7%) remained the most dominant bacteria, followed by bacteria belonging to Rikenellaceae RC9 (19.6%), Fibrobacter (9.9%), Prevotellaceae UCG003 (4.2%), and Succinivibrionaceae UCG002 (4.0%), accounting for 58% of the overall bacterial composition. After 48 h of incubation, members of the genus Prevotella 1 (18.1%) remained the most dominant bacteria, followed by bacteria belonging to Rikenellaceae RC9 (15.8%), Fibrobacter (12.6%), Ruminococcaceae UCG010 (5.5%), F082 (Bacteroidales, 5.0%), and Christensenellaceae R7 (4.7%), accounting for 62% of the overall bacterial composition.

The composition of the bacterial community attached to reeds in the rumen of Tarim red deer during the incubation process. A The bacterial composition at the genus level after incubating reeds for 1, 6, 12 and 48 h in the rumen of Tarim red deer. PCoA results based on unweighted unifrac distances (B) and weighted distances (C). Canonical correlation analysis (D) showing the changes in the bacteria attached to reeds across four time points. Black circles indicate the representative taxa at each time point
PCoA based on unweighted unifrac (Fig. 2B) and weighted unifrac (Fig. 2C) distance metrics showed that the composition and structure of the bacterial community attached to reeds at 48 h were different from those observed at the other three time points. Moreover, CCA was used to identify the possible taxa that contributed to the differences in the community. A total of 18 bacterial taxa were associated with the four incubation times (Fig. 2D).Then, we compared the relative abundances of these taxa across four time points. The results showed that the abundances of bacteria attached to reeds, including the members of Succinivibrionaceae UCG002, Sphaerochaeta, Succinivibrio, and Acinetobacter, decreased after 1 h of incubation in the rumen. The relative abundances of bacteria belonging to Christensenellaceae R7, Rikenellaceae RC9, Prevotellaceae UCG001, Ruminococcaceae UCG010, Treponema 2, Succiniclasticum, Papillibacter, Desulfovibrio, and Anaeroplasma increased after 48 h of incubation compared the abundances observed 1 h after incubation (Table 2). In addition, the proportions of Prevotella 1, Prevotellaceae UCG003, and Alloprevotella decreased linearly, and that of Fibrobacter spp. increased linearly, with increasing incubation time (Table 2).
Dynamics of the bacterial communities attached to the cottonseed hulls over time
A total of 18 phyla were identified based on the 3576 OTUs in the cottonseed hulls at four time points (Figure S2).The phyla Bacteroidetes, Firmicutes, and Proteobacteria were the dominant bacteria, accounting for more than 89% of the OTUs detected during the incubation of cottonseed hulls in the rumen of Tarim red deer. The relative abundances of Bacteroidetes (1 h = 73.6%, 6 h = 71.8%, 12 h = 66.3%, 48 h = 61.9%, respectively) and Proteobacteria (1 h = 7.0%, 6 h = 3.9%, 12 h = 3.1%, 48 h = 2.0%, respectively) decreased linearly with increasing incubation time, whereas the proportion of Firmicutes increased linearly (1 h = 8.9%, 6 h = 17.8%, 12 h = 20.5%, 48 h = 25.3%, respectively).
At the genus level (Fig. 3A), members of the genus Prevotella 1 (34.5%) were the most dominant bacteria after 1 h of incubation of cottonseed hulls in the rumen of Tarim red deer, followed by bacteria belonging to Rikenellaceae RC9 (18.4%), Bacteroidales RF16 (10.6%), Succinivibrio (6.0%), Sphaerochaeta (5.4%), and Prevotellaceae UCG003 (5.0%), accounting for approximately 80% of the overall bacterial composition. After 6 h of incubation, the members of the genus Prevotella 1 (40.9%) remained the most dominant bacteria, followed by bacteria belonging to Rikenellaceae RC9 (14.2%), Christensenellaceae R7 (6.7%), Bacteroidales RF16 (5.8%), and Prevotellaceae UCG001 (3.5%), accounting for approximately 76% of the overall bacterial composition. After 12 h of incubation, members of the genus Prevotella 1 (35.9%) remained the most dominant bacteria, followed by bacteria belonging to Rikenellaceae RC9 (14.2%), Christensenellaceae R7 (6.3%), Bacteroidales RF16 (4.2%), Treponema 2 (4.0%), and Prevotellaceae UCG003 (3.1%), accounting for approximately 68% of the overall bacterial composition (Fig. 3A). After 48 h of incubation, members of the genus Prevotella 1 (25.8%) remained the most dominant bacteria, followed by bacteria belonging to Rikenellaceae RC9 (22.0%), Butyrivibrio 2 (5.5%), Treponema 2 (4.8%), and Christensenellaceae R7 (3.5%), accounting for 69.6% of the overall bacterial composition.

The composition of the bacterial community attached to cottonseed hulls in the rumen of Tarim red deer during the incubation process. A The bacterial composition at the genus level after incubation of cottonseed hulls for 1, 6, 12 and 48 h in the rumen of Tarim red deer. PCoA results based on unweighted unifrac distances (B) and weighted distances (C). Canonical correlation analysis (D) showing the changes in the bacteria attached to cottonseed hulls across four time points. Black circles indicate the representative taxa at each time point
The PCoA results revealed that the composition of the bacterial community attached to the cottonseed hulls at 48 h was clearly different from that at the other incubation time points based on unweighted unifrac distances (Fig. 3B) and weighted unifrac distances (Fig. 3C), explaining at least 45% of the variation. We also applied CCA to identify the taxa representing each time point. A total of 18 bacterial taxa were identified as being associated with the four different time points (Fig. 3D), the relative abundances of which were further compared (Table 3). The relative abundances of members of Butyrivibrio 2, Treponema 2, Lachnospiraceae FCS020, and Ruminococcaceae NK4A214 increased linearly from 1 to 48 h. The proportions of members of Prevotella 1 and Prevotellaceae UCG001 were lower at 48 h than at the other three incubation time points, whereas those of Prevotellaceae Ga6A1, Succiniclasticum, and Rikenellaceae RC9 were higher at 48 h than at the other time points. The proportions of the members of Christensenellaceae R7, Lachnospiraceae XPB1014, and Fibrobacter increased after 1 h of incubation in the rumen, while the relative abundances of Prevotellaceae UCG003, Sphaerochaeta, Succinivibrio, and Ruminococcaceae UCG010 decreased (Table 3).
Discussion
In the present study, the results showed that the taxa Prevotella 1 and Rikenellaceae RC9 were the mostly widely distributed bacteria attached to both reeds (33–51%) and cottonseed hulls (47–55%) at all the incubation times (Figs. 2A, 3B), which is consistent with findings regarding the rumen microbiota of ruminants (Henderson et al. 2015), and with the results of our previous comparison of the rumen microbiota of cattle, sheep and Tarim red deer (Qian et al. 2017), which exhibited a high abundance of Prevotella spp. Interestingly, Prevotella spp. were also documented to be predominant bacteria in the rumen of elk, white-tailed deer, roe deer, sika deer, reindeer and moose (Gruninger et al. 2014; Ishaq and Wright 2014; Li et al. 2013, 2014; Sundset et al. 2007). These results suggest that the host phylogeny strongly affects the microbiota in the gastrointestinal tract (Amato et al. 2018), which may be a result of co-evolution (Kohl et al. 2018; Moeller et al. 2013). Prevotella spp. represent one of the most abundant genera in the rumen, exhibiting genetic and metabolic diversity (Bekele et al. 2010; Purushe et al. 2010), and playing major roles in carbohydrate metabolism, such as degradation of hemicellulose, starch, xylan, lignin and pectin (Cotta 1992; Dehority 1966; Gardner et al. 1995; Kabel et al. 2011), and nitrogen metabolism (Kim et al. 2017; Stevenson and Weimer 2007). A previous study demonstrated that members of Rikenellaceae RC9 are closely related to members of the genus Alistipes within the family Rikenellaceae (Seshadri et al. 2018), which play possible roles in degrading plant-derived polysaccharides (He et al. 2015; Peng et al. 2015). Taken together, these findings suggest that the members of Prevotella 1 and Rikenellaceae RC9 are involved in the fermentation of carbohydrates and nitrogen in the rumen of Tarim red deer.
The present results illustrated that the diversity changed over incubation time in both reeds and cottonseed hulls (Fig. 1), indicating the dynamics of the rumen microbiota attached to the forage. However, we observed that the microbial diversity decreased from 1 to 12 h in the reed group, whereas an increasing trend was observed in the cottonseed hull group, indicating that the colonisation events between reeds and cottonseed hulls were distinct. This finding is consistent with the previous observation that the composition and structure of the attached microbiota between rice straw and alfalfa differed significantly in the rumen of dairy cattle (Liu et al. 2016), which may be caused by different chemical ingredients, such as condensed tannin (Yu et al. 1995), and available nutrients, such as easily fermentable carbohydrates (Huws et al. 2016). Indeed, the DM content in reed (88%) was lower than that in cottonseed hulls (98%). On the other hand, this discrepancy in colonisation partially demonstrated that the different forage types affected the rumen microbiota (Pitta et al. 2014), likely due to differences in successional colonisation during rumen fermentation.
We further identified the different functional microbes that were present during colonisation between reeds and cottonseed hulls. The abundance of the phylum Proteobacteria was lower at 48 h of incubation than at 1 h of incubation (Figures S1 and S2), which is consistent with the results observed upon incubation of rice straw and alfalfa in the rumen of cattle (Liu et al. 2016). The phylum Proteobacteria has been identified in the rumen tissue of pre-weaning calves, suggesting a role for these bacteria in the scavenging of oxygen, facilitating microbiome colonisation (Malmuthuge et al. 2014). Therefore, the increase in Proteobacteria abundance may have been the result of oxygen infusion through the rumen cannula.
The present study showed that the relative abundance of Fibrobacter, Treponema 2, Ruminococcaceae NK4A214, and Succiniclasticum increased with incubation time in both reeds and cottonseed hulls (Tables 2, 3, Figures S3 and S4), which is partially consistent with the colonisation of rice straw and alfalfa (Liu et al. 2016), wheat straw (Jin et al. 2018) and switch grass (Piao et al. 2014). Fibrobacteria spp. are an important group of bacteria that play a primary role in the degradation of cellulosic plant biomass in the rumen (Abdul Rahman et al. 2016; Béra-Maillet et al. 2004; Tajima et al. 2001). The genus Treponema, a member of the phylum Spirochetes, is always detected in the gastrointestinal tracts of ruminants; these bacteria contain a wide variety of carbohydrate-active enzymes, as suggested by genome analysis (Rosewarne et al. 2012), and are reported to be associated with the degradation of cellulose and pectin (Liu et al. 2015; Stanton and Canale-Parola 1980). Members of the Ruminococcaceae family are important for the degradation of plant fibres, as demonstrated by metagenomic (Kim et al. 2011) and transcriptomic analyses (Christopherson et al. 2014), and the abundances of these species were positively correlated with NDF content in cattle rumen (Liu et al. 2016). Succiniclasticum species are common inhabitants in the rumens of pasture-fed cows, converting succinate to propionate as the sole energy-yielding mechanism (van Gylswyk 1995). Moreover, the NDF content was dramatically decreased over the incubation time in both reed and cottonseed hulls (Table 1). Together, these results suggest that these enriched bacteria play an important role in forage degradation.
In contrast, we also found that the abundance of the genus Prevotella1 decreased in both groups, which is consistent with previous results obtained by incubation of rice straw and alfalfa in the rumen of cattle (Liu et al. 2016), and ryegrass in rumen of cows (Huws et al. 2016). Although we do not know the metabolic function of Prevotella 1, we found that this group of bacteria are similar to Prevotella copri (93% similarity) by aligning the OTU sequence to the NCBI sequence database. Previous studies have demonstrated that P. copri contains a number of enzymes and gene clusters essential for the fermentation and utilisation of complex polysaccharides (Dodd et al. 2011) and could play a potential role in glucose metabolism (Kovatcheva-Datchary et al. 2015). In addition, Prevotella spp. have diverse activities that degrade the hemicellulose matrix formed by pectins, hemicellulose and peptides (Rubino et al. 2017). Moreover, the relative abundance of Prevotella spp. in the rumen of Hu sheep decreased when the forage changed from alfalfa hay to corn stover, which partially resulted from the low content of neutral detergent-soluble and crude protein in corn stover (Xie et al. 2018).Therefore, the decrease in abundance of the genus Prevotella 1 was likely a result of a decrease in utilisable soluble nutrients during incubation, such as NDF and CP in both reed and cottonseed hulls (Table 1; Huws et al. 2016; Liu et al. 2016).
We also found that the bacteria attached to reeds and cottonseed hulls differed during the incubation process. For instance, the relative abundances of Ruminococcaceae UCG 010 and Papillibacter first decreased and then increased in the reed group, and the proportion of Sphaerochaeta and Erysipelotrichaceae UCG 004 also exhibited a similar trend in the cottonseed hull group (Tables 2, 3, Figures S3 and S4). Moreover, the relative abundances of Butyrivibrio 2 and Lachnospiraceae FCS020 increased in only the cottonseed hull group (Table 3). These findings further indicated that the forage type affected the colonisation events. Similar dynamics were also observed during the incubation of ryegrass in the cow rumen (Huws et al. 2016). These results further indicated the different colonisation stages during the degradation of plant fibres, which may be associated with changes in the rumen ecological niches and bacterial diversity (Rubino et al. 2017). The abundance of Papillibacter spp. was reported to be reduced in the rumen of cattle when the amount of dietary concentrate was increased (Mao et al. 2016); these bacteria grow on a limited range of aromatic compounds and crotonate but not carbohydrates, organic acids or alcohols, resulting in the production of acetate (Defnoun et al. 2000). The members of the family Ruminococcaceae are important not only in fibre degradation, as discussed above, but also in butyrate production (Bui et al. 2016). Interestingly, we found that Ruminococcaceae UCG 010 was similar to Intestinimonas butyriciproducens (93–100% similarity), which can produce butyrate via the acetyl-CoA pathway and the glutamate, succinate and lysine pathways, in which amino acids such lysine and glutamate act as substrates (Bui et al. 2015; Kläring et al. 2013). Butyrate serves as a major source of metabolic energy in ruminants and as a host signal (Bergman 1990; Hamer et al. 2008). Together, these results suggest that Papillibacter spp. and Ruminococcaceae UCG 010 may be responsible for the degradation of chemical components in reeds, leading to the production of acetate and butyrate.
In the cottonseed hull group, the relative abundance of Sphaerochaeta spp. and Erysipelotrichaceae UCG 004 increased after 48 h of incubation. The genus Sphaerochaeta is a member of the phylum Spirochaetes, utilising hydrolysis products of plant polymers as substrates, such as xylan and pectin, but not cellulose and amino acids (Paster and Canale-Parola 1982). Furthermore, genome analysis revealed that Sphaerochaeta species are highly enriched in fermentation and carbohydrate metabolism genes relative to other spirochetes, indicating a fermentative lifestyle (Caro-Quintero et al. 2012), and were highly abundant in the rumen of sheep fed corn stover (Xie et al. 2018). On the other hand, Erysipelotrichaceae UCG 004 was found by sequence comparison to be related to Turicibacter sanguinis (84%). The latter utilised only maltose and 5-ketogluconate as carbohydrate sources, producing lactate, but this species did not reduce nitrate (Bosshard et al. 2002). Interestingly, the abundance of Sphaerochaeta spp. increased in steers fed 1% nitrate, which was reduced to ammonia (Zhao et al. 2015). Together, these findings suggest the possible cooperation between members of Sphaerochaeta and Erysipelotrichaceae UCG 004 in the rumen of the cottonseed hull group, facilitating the efficient degradation of plant fibres.
In summary, our findings showed the temporal colonisation of bacteria attached to reed sand cottonseed hulls in the rumen of Tarim red deer living in the Taklimakan desert. The results showed that there were common bacteria attached to the different forages. However, differing dynamics of bacterial colonisation were also observed between the reeds and cottonseed hulls in the rumen. In addition to being important for improving the efficiency of forage nutrients, these findings also improve our knowledge regarding the interactions between plants and the rumen microbiota of desert ruminants, which may provide insights for the discovery of novel enzymes for cellulosic biomass production.
References
Abdul Rahman N, Parks D, Vanwonterghem I, Morrison M, Tyson GW, Hugenholtz P (2016) A phylogenomic analysis of the bacterial phylum Fibrobacteres. Front Microbiol. https://doi.org/10.3389/fmicb.2015.01469
Amato KR et al (2018) Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J 1:2. https://doi.org/10.1038/s41396-018-0175-0
AOAC (2000) Official methods of analysis, 17th edn. AOAC, Gaithersburg
Bekele AZ, Koike S, Kobayashi Y (2010) Genetic diversity and diet specificity of ruminal Prevotella revealed by 16S rRNA gene-based analysis. FEMS Microbiol Lett 305:49–57. https://doi.org/10.1111/j.1574-6968.2010.01911.x
Béra-Maillet C, Ribot Y, Forano E (2004) Fiber-degrading systems of different strains of the genus Fibrobacter. Appl Environ Microbiol 70:2172–2179. https://doi.org/10.1128/aem.70.4.2172-2179.2004
Bergman EN (1990) Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev 70:567–590
Bosshard PP, Zbinden R, Altwegg M (2002) Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, Gram-positive bacterium. Int J Syst Evol Microbiol 52:1263–1266. https://doi.org/10.1099/00207713-52-4-1263
Brook SM, Donnithorne-Tait D, Lorenzini R, Lovari S, Masseti M, Pereladova O, Ahmad K, Thakur M (2017) Cervus hanglu (amended version of 2017 assessment). The IUCN Red list of threatened species 2017 e.T4261A120733024. https://doi.org/10.2305/IUCN.UK.2017-2.RLTS.T113259123A113281791.en
Bui TPN, Ritari J, Boeren S, de Waard P, Plugge CM, de Vos WM (2015) Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat Commun 6:10062. https://doi.org/10.1038/ncomms10062
Bui TPN, Shetty SA, Lagkouvardos I, Ritari J, Chamlagain B, Douillard FP, Paulin L, Piironen V, Clavel T, Plugge CM, de Vos WM (2016) Comparative genomics and physiology of the butyrate-producing bacterium Intestinimonas butyriciproducens. Environ Microbiol Rep 8:1024–1037. https://doi.org/10.1111/1758-2229.12483
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R (2010a) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. https://doi.org/10.1093/bioinformatics/btp636
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI (2010b) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624
Caro-Quintero A, Ritalahti KM, Cusick KD, Löffler FE, Konstantinidis KT (2012) The chimeric genome of sphaerochaeta: nonspiral spirochetes that break with the prevalent dogma in spirochete biology. mBio. https://doi.org/10.1128/mbio.00025-12
Christopherson MR, Dawson JA, Stevenson DM, Cunningham AC, Bramhacharya S, Weimer PJ, Kendziorski C, Suen G (2014) Unique aspects of fiber degradation by the ruminal ethanologen Ruminococcus albus 7 revealed by physiological and transcriptomic analysis. BMC Genom 15:1066. https://doi.org/10.1186/1471-2164-15-1066
Cotta M (1992) Interaction of ruminal bacteria in the production and utilization of maltooligosaccharides from starch. Appl Environ Microbiol 58:48–54
De Mulder T, Goossens K, Peiren N, Vandaele L, Haegeman A, De Tender C, Ruttink T, de Wiele TV, De Campeneere S (2017) Exploring the methanogen and bacterial communities of rumen environments: solid adherent, fluid and epimural. FEMS Microbiol Ecol. https://doi.org/10.1093/femsec/fiw251
Defnoun S, Labat M, Ambrosio M, Garcia JL, Patel BK (2000) Papillibacter cinnamivorans gen. nov., sp. nov., a cinnamate-transforming bacterium from a shea cake digester. Int J Syst Evol Microbiol 50:1221–1228. https://doi.org/10.1099/00207713-50-3-1221
Dehority BA (1966) Characterization of several bovine rumen bacteria isolated with a xylan medium. J Bacteriol 91:1724–1729
Ding LM, Lascano GJ, Heinrichs AJ (2015) Effect of precision feeding high- and low-quality forage with different rumen protein degradability levels on nutrient utilization by dairy heifers. J Anim Sci 93:3066–3075. https://doi.org/10.2527/jas.2014-8260
Dodd D, Mackie RI, Cann IKO (2011) Xylan degradation, a metabolic property shared by rumen and human colonic Bacteroidetes. Mol Microbiol 79:292–304. https://doi.org/10.1111/j.1365-2958.2010.07473.x
Dufrene M, Legendre P (1997) Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol Monogr 67:345–366. https://doi.org/10.2307/2963459
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Eisler MC, Lee MR, Tarlton JF, Martin GB, Beddington J, Dungait JA, Greathead H, Liu J, Mathew S, Miller H, Misselbrook T, Murray P, Vinod VK, Van Saun R, Winter M (2014) Agriculture: steps to sustainable livestock. Nature 507:32–34
Gardner RG, Wells JE, Russell JB, Wilson DB (1995) The cellular location of Prevotella ruminicola beta-1,4-D-endoglucanase and its occurrence in other strains of ruminal bacteria. Appl Environ Microbiol 61:3288–3292
Gruninger RJ, Sensen CW, McAllister TA, Forster RJ (2014) Diversity of rumen bacteria in Canadian cervids. PLoS One 9:e89682. https://doi.org/10.1371/journal.pone.0089682
Hamer HM, Jonkers D, Venema K, Vanhoutvinn S, Troost FJ, Brummer RJ (2008) Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 27:104–119. https://doi.org/10.1111/j.1365-2036.2007.03562.x
He B, Nohara K, Ajami NJ, Michalek RD, Tian X, Wong M, Losee-Olson SH, Petrosino JF, Yoo SH, Shimomura K, Chen Z (2015) Transmissible microbial and metabolomic remodeling by soluble dietary fiber improves metabolic homeostasis. Sci Rep 5:10604. https://doi.org/10.1038/srep10604
Henderson G, Cox F, Ganesh S, Jonker A, Young W, Janssen PH (2015) Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep 5:14567. https://doi.org/10.1038/srep14567
Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G, Luo S, Clark DS, Chen F, Zhang T, Mackie RI, Pennacchio LA, Tringe SG, Visel A, Woyke T, Wang Z, Rubin EM (2011) Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331:463–467. https://doi.org/10.1126/science.1200387
Huws SA, Mayorga OL, Theodorou MK, Onime LA, Kim EJ, Cookson AH, Newbold CJ, Kingston-Smith AH (2013) Successional colonization of perennial ryegrass by rumen bacteria. Lett Appl Microbiol 56:186–196. https://doi.org/10.1111/lam.12033
Huws SA, Edwards JE, Creevey CJ, Rees Stevens P, Lin W, Girdwood SE, Pachebat JA, Kingston-Smith AH (2016) Temporal dynamics of the metabolically active rumen bacteria colonizing fresh perennial ryegrass. FEMS Microbiol Ecol. https://doi.org/10.1093/femsec/fiv137
Ishaq SL, Wright ADG (2014) High-throughput DNA sequencing of the ruminal bacteria from Moose (Alces alces) in Vermont, Alaska, and Norway. Microb Ecol 68:185–195. https://doi.org/10.1007/s00248-014-0399-0
Jin W, Wang Y, Li Y, Cheng Y, Zhu W (2018) Temporal changes of the bacterial community colonizing wheat straw in the cow rumen. Anaerobe 50:1–8. https://doi.org/10.1016/j.anaerobe.2018.01.004
Kabel MA, Yeoman CJ, Han Y, Dodd D, Abbas CA, de Bont JA, Morrison M, Cann IK, Mackie RI (2011) Biochemical characterization and relative expression levels of multiple carbohydrate esterases of the xylanolytic rumen bacterium Prevotella ruminicola 23 grown on an ester-enriched substrate. Appl Environ Microbiol 77:5671–5681. https://doi.org/10.1128/AEM.05321-11
Kim H, Lee I, Kwon Y, Kim BC, Ha S, Lee J-H, Kim J (2011) Immobilization of glucose oxidase into polyaniline nanofiber matrix for biofuel cell applications. Biosens Bioelectron 26:3908–3913. https://doi.org/10.1016/j.bios.2011.03.008
Kim JN, Méndez-García C, Geier RR, Iakiviak M, Chang J, Cann I, Mackie RI (2017) Metabolic networks for nitrogen utilization in Prevotella ruminicola 23. Sci Rep 7:7851. https://doi.org/10.1038/s41598-017-08463-3
Kläring K, Hanske L, Bui N, Charrier C, Blaut M, Haller D, Plugge CM, Clavel T (2013) Intestinimonas butyriciproducens gen. nov., sp. nov., a butyrate-producing bacterium from the mouse intestine. Int J Syst Evol Microbiol 63:4606–4612. https://doi.org/10.1099/ijs.0.051441-0
Kohl KD, Denise Dearing M, Bordenstein SR (2018) Microbial communities exhibit host species distinguishability and phylosymbiosis along the length of the gastrointestinal tract. Mol Ecol 27:1874–1883. https://doi.org/10.1111/mec.14460
Kononoff PJ, Heinrichs AJ (2003) The effect of corn silage particle size and cottonseed hulls on cows in early lactation. J Dairy Sci 86:2438–2451. https://doi.org/10.3168/jds.S0022-0302(03)73838-7
Kovatcheva-Datchary P, Nilsson A, Akrami R, Lee Ying S, De Vadder F, Arora T, Hallen A, Martens E, Björck I, Bäckhed F (2015) Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of Prevotella. Cell Metab 22:971–982. https://doi.org/10.1016/j.cmet.2015.10.001
Larue R, Yu Z, Parisi VA, Egan AR, Morrison M (2005) Novel microbial diversity adherent to plant biomass in the herbivore gastrointestinal tract, as revealed by ribosomal intergenic spacer analysis and RRS gene sequencing. Environ Microbiol 7:530–543. https://doi.org/10.1111/j.1462-2920.2005.00721.x
Li ZP, Liu HL, Li GY, Bao K, Wang KY, Xu C, Yang YF, Yang FH, Wright ADG (2013) Molecular diversity of rumen bacterial communities from tannin-rich and fiber-rich forage fed domestic Sika deer (Cervus nippon) in China. BMC Microbiol 13:151. https://doi.org/10.1186/1471-2180-13-151
Li ZP, Zhang ZG, Xu C, Zhao JB, Liu HL, Fan ZY, Yang F, Wright ADG, Li GY (2014) Bacteria and methanogens differ along the gastrointestinal tract of Chinese roe deer (Capreolus pygargus). PLoS One 9:e114513. https://doi.org/10.1371/journal.pone.0114513
Liu J, Pu YY, Xie Q, Wang JK, Liu JX (2015) Pectin induces an in vitro rumen microbial population shift attributed to the pectinolytic Treponema group. Curr Microbiol 70:67–74. https://doi.org/10.1007/s00284-014-0672-y
Liu J, Zhang M, Xue C, Zhu W, Mao S (2016) Characterization and comparison of the temporal dynamics of ruminal bacterial microbiota colonizing rice straw and alfalfa hay within ruminants. J Dairy Sci 99:9668–9681. https://doi.org/10.3168/jds.2016-11398
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R (2011) UniFrac: an effective distance metric for microbial community comparison. ISME J 5:169
Malmuthuge N, Griebel PJ, Guan LL (2014) Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Appl Environ Microbiol 80:2021–2028. https://doi.org/10.1128/aem.03864-13
Mao SY, Huo WJ, Zhu WY (2016) Microbiome-metabolome analysis reveals unhealthy alterations in the composition and metabolism of ruminal microbiota with increasing dietary grain in a goat model. Environ Microbiol 18:525–541. https://doi.org/10.1111/1462-2920.12724
McAllister TA, Bae HD, Jones GA, Cheng KJ (1994) Microbial attachment and feed digestion in the rumen. J Anim Sci 72:3004–3018
Moeller AH, Peeters M, Ndjango J-B, Li Y, Hahn BH, Ochman H (2013) Sympatric chimpanzees and gorillas harbor convergent gut microbial communities. Genome Res 23:1715–1720. https://doi.org/10.1101/gr.154773.113
Paster BJ, Canale-Parola E (1982) Physiological diversity of rumen spirochetes. Appl Environ Microbiol 43:686–693
Peng B, Huang S, Liu T, Geng A (2015) Bacterial xylose isomerases from the mammal gut Bacteroidetes cluster function in Saccharomyces cerevisiae for effective xylose fermentation. Microb Cell Factories 14:70. https://doi.org/10.1186/s12934-015-0253-1
Piao H, Lachman M, Malfatti S, Sczyrba A, Knierim B, Auer M, Tringe SG, Mackie RI, Yeoman CJ, Hess M (2014) Temporal dynamics of fibrolytic and methanogenic rumen microorganisms during in situ incubation of switchgrass determined by 16S rRNA gene profiling. Front Microbiol. https://doi.org/10.3389/fmicb.2014.00307
Pitta DW, Kumar S, Veiccharelli B, Parmar N, Reddy B, Joshi CG (2014) Bacterial diversity associated with feeding dry forage at different dietary concentrations in the rumen contents of Mehshana buffalo (Bubalus bubalis) using 16S pyrotags. Anaerobe 25:31–41. https://doi.org/10.1016/j.anaerobe.2013.11.008
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650. https://doi.org/10.1093/molbev/msp077
Purushe J, Fouts D, Morrison M, White B, Mackie R, Coutinho P, Henrissat B, Nelson K (2010) Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb Ecol 60:721–729. https://doi.org/10.1007/s00248-010-9692-8
Qian WX, Li ZP, Ao WP, Zhao GY, Li GY, Wu JP (2017) Bacterial community composition and fermentation in the rumen of Xinjiang brown cattle (Bos taurus), Tarim red deer (Cervus elaphus yarkandensis), and Karakul sheep (Ovis aries). Can J Microbiol 63:375–383. https://doi.org/10.1139/cjm-2016-0596
Qiao J, Yang W, Gao X (2006) Natural diet and food habitat use of the Tarim red deer, Cervus elaphus yarkandensis. Chin Sci Bull 51:147–152. https://doi.org/10.1007/s11434-006-8219-7
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. https://doi.org/10.1093/nar/gks1219
Rosewarne CP, Cheung JL, Smith WJM, Evans PN, Tomkins NW, Denman SE, Cuiv PO, Morrison M (2012) Draft genome sequence of Treponema sp strain JC4, a novel spirochete isolated from the bovine rumen. J Bacteriol 194:4130. https://doi.org/10.1128/Jb.00754-12
Rubino F, Carberry C, Waters SM, Kenny D, McCabe MS, Creevey CJ (2017) Divergent functional isoforms drive niche specialisation for nutrient acquisition and use in rumen microbiome. ISME J 11:932–944. https://doi.org/10.1038/ismej.2016.172
Russell JB, Rychlik JL (2001) Factors that alter rumen microbial ecology. Science 292:1119–1122
Seshadri R et al (2018) Cultivation and sequencing of rumen microbiome members from the Hungate1000 collection. Nat Biotechnol 1:2. https://doi.org/10.1038/nbt.4110
Stanton TB, Canale-Parola E (1980) Treponema bryantii sp. nov., a rumen spirochete that interacts with cellulolytic bacteria. Arch Microbiol 127:145–156
Stevenson DM, Weimer PJ (2007) Dominance of prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl Microbiol Biotechnol 75:165–174. https://doi.org/10.1007/s00253-006-0802-y
Sundset MA, Praesteng KE, Cann IK, Mathiesen SD, Mackie RI (2007) Novel rumen bacterial diversity in two geographically separated sub-species of reindeer. Microb Ecol 54:424–438. https://doi.org/10.1007/s00248-007-9254-x
Svartström O, Alneberg J, Terrapon N, Lombard V, de Bruijn I, Malmsten J, Dalin A-M, El Muller E, Shah P, Wilmes P, Henrissat B, Aspeborg H, Andersson AF (2017) Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. ISME J 11:2538. https://doi.org/10.1038/ismej.2017.108
Tajima K, Aminov RI, Nagamine T, Matsui H, Nakamura M, Benno Y (2001) Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl Environ Microbiol 67:2766–2774. https://doi.org/10.1128/AEM.67.6.2766-2774.2001
Tumur A, Abliz D, Halik M (2013) Habitat dynamics and its influence on the genetic diversity of Tarim red deer (Cervus elaphus yarkandensis) Xayar population of Xinjiang, China. Quat Int 311:140–145. https://doi.org/10.1016/j.quaint.2013.07.007
van Gylswyk NO (1995) Succiniclasticum ruminis gen. nov., sp. nov., a ruminal bacterium converting succinate to propionate as the sole energy-yielding mechanism. Int J Syst Bacteriol 45:297–300. https://doi.org/10.1099/00207713-45-2-297
Van Soest PJ, Robertson JB, Lewis BA (1991) Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J Dairy Sci 74:3583–3597. https://doi.org/10.3168/jds.S0022-0302(91)78551-2
Xie X, Yang C, Guan LL, Wang J, Xue M, Liu JX (2018) Persistence of cellulolytic bacteria Fibrobacter and Treponema after short-term corn stover-based dietary intervention reveals the potential to improve rumen fibrolytic function. Front Microbiol. https://doi.org/10.3389/fmicb.2018.01363
Yu F, McNabb WC, Barry TN, Waghorn GC (1995) Effect of condensed tannin in cottonseed hulls upon the in vitro degradation of cottonseed kernel proteins by rumen microorganisms. J Sci Food Agric 69:223–234. https://doi.org/10.1002/jsfa.2740690213
Zhao L, Meng Q, Ren L, Liu W, Zhang X, Huo Y, Zhou Z (2015) Effects of nitrate addition on rumen fermentation, bacterial biodiversity and abundance. Asian-Australas J Anim Sci 28:1433–1441. https://doi.org/10.5713/ajas.15.0091
Acknowledgements
This study is supported by the Opening Project of Key Laboratory of Tarim Animal Husbandry Science and Technology, Xinjiang Production and Construction Group (Grant No. HS201702) to ZPL, and National Natural Science Foundation of China (Grant Nos. 31460610 and 31660671) to WXQ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare no conflict of interest exits among the authors, and manuscript is approved by all authors for submission. All authors declare that the work is original research that has not been published previously, and is not under consideration for publication elsewhere.
Human and animal participants
All animal care procedures were approved and authorised by the animal ethics committee of Tarim University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
. The dynamic composition at the phylum level of bacteria attached to reeds after 1, 6, 12, and 48 h of incubation in the rumen of Tarim red deer. (TIFF 779 kb)
Figure S2
. The dynamic composition at the phylum level of bacteria attached to cottonseed hulls after 1, 6, 12, and 48 h of incubation in the rumen of Tarim red deer. (TIFF 731 kb)
Figure S3. Taxonomic representation showing the statistically and biologically difference in reed after 1, 6, 12, and 48
h of incubation in the rumen of Tarim red deer. Differences are represented by the colour of the most abundant class (Green, purple, red and blue indicating the samples after 1, 6, 12 and 48 h). The diameter of each circle’s diameter is proportional to the taxa abundance. (TIFF 1027 kb)
Figure S4. Taxonomic representation showing the statistically and biologically difference in cottonseed hulls over the incubation time.
Differences are represented by the colour of the most abundant class (Green, purple, red and blue indicating the samples after 1, 6, 12 and 48 h). The diameter of each circle’s diameter is proportional to the taxa abundance. (TIFF 1225 kb)
Rights and permissions
About this article

Cite this article
Qian, W., Ao, W., Jia, C. et al. Bacterial colonisation of reeds and cottonseed hulls in the rumen of Tarim red deer (Cervus elaphus yarkandensis). Antonie van Leeuwenhoek 112, 1283–1296 (2019). https://doi.org/10.1007/s10482-019-01260-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-019-01260-0