
Abstract
The taxonomic position of strains Ab112T (CBAS 572T) and Ab227_MC (CBAS 573) was evaluated by means of genomic taxonomy. These isolates represent the dominant flora cultured from the healthy marine sponge Arenosclera brasiliensis, endemic to Rio de Janeiro. Strains CBAS 572T and CBAS 573 shared >98 % 16S rRNA sequence identity with Endozoicomonas numazuensis and Endozoicomonas montiporae. In silico DNA–DNA Hybridization, i.e. genome-to-genome distance (GGD), amino acid identity (AAI) and average nucleotide identity (ANI) further showed that these strains had <70 %, at maximum 71.1 and 78 % of identity, respectively, to their closest neighbours E. numazuensis and E. montiporae. The DNA G+C content of CBAS 572T and CBAS 573 were 47.6 and 47.7 mol%, respectively. Phenotypic and chemotaxonomic features also allowed a separation from the type strains of their phylogenetic neighbours. Useful phenotypic features for discriminating CBAS 572T and CBAS 573 from E. numazuensis and E. montiporae species include C8 esterase, N-acetyl-β-glucosaminidase, citric acid, uridine and siderophore. The species Endozoicomonas arenosclerae sp. nov. is proposed to harbour the new isolates. The type strain is CBAS 572T (=Ab112T).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Endozoicomonas belongs to the order Oceanospirillales of the class Gammaproteobacteria, and comprises six species: Endozoicomonas elysicola (Kurahashi and Yokota 2007), Endozoicomonas montiporae (Yang et al. 2010), Endozoicomonas numazuensis (Nishijima et al. 2013), Endozoicomonas euniceicola; Endozoicomonas gorgoniicola (Pike et al. 2013), and Endozoicomonas atrinae (Hyun et al. 2014). Endozoicomonas spp. are found in marine environments, including healthy and diseased sponges (Neave et al. 2014; Mohamed et al. 2008; Nishijima et al. 2013), corals (Yang et al. 2010; Pike et al. 2013), ascidians (Martínez-García et al. 2007), nudibranchs (Kurahashi and Yokota 2007), polychaetes (Goffredi et al. 2007), sea anemones (Du et al. 2010), starfishes (Choi et al. 2010) and bivalves (Zielinski et al. 2009; Hyun et al. 2014). Novel Endozoicomonas isolates related to E. numazuensis were found to be dominantly present among the culturable microbiome of healthy marine sponge Arenosclera brasiliensis which is endemic from Rio de Janeiro (Rua et al. 2014). However, it was inappropriate to allocate the novel isolates to a known Endozoicomonas species. Several of these novel Endozoicomonas isolates had strong antimicrobial activity against Gram-positive Bacillus subtilis.
Genomic taxonomy has already been successfully applied as an alternative to the traditional species description and re-classification (Thompson et al. 2009; Haley et al. 2010; Thompson et al. 2011a, 2013b; Moreira et al. 2014; Thompson et al. 2014). For example, the genus Listonella was reclassified as a later heterotypic synonym of the genus Vibrio (Thompson et al. 2011b), and a new taxonomic framework for the genus Prochlorococcus was proposed with the genomic descriptions of new species (Thompson et al. 2013c). Thus, the aim of the present study was to determine the taxonomic position of the novel isolates CBAS 572T and CBAS 573 using whole-genome-based taxonomic analysis (Thompson et al. 2014).
Materials and methods
The isolation of Endozoicomonas strains was performed as previously described (Rua et al. 2014). The representative isolates Ab112T and Ab227_MC are deposited in the Bacteria Collection of Environmental and Health (CBAS) at Oswaldo Cruz Institute (IOC), FIOCRUZ (Rio de Janeiro, Brazil) (http://cbas.fiocruz.br/) under the accession numbers CBAS 572T and CBAS 573, respectively.
Genome sequencing and analysis
Genomic DNA was extracted using the method of Pitcher et al. (1989) and the DNA libraries were built using the Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA, USA). The size distribution of the libraries was evaluated using the 2100 Bioanalyzer and the High Sensitivity DNA kit (Agilent, Santa Clara, CA, USA). The accurate quantification of the libraries was achieved using the 7500 Real Time PCR (Applied Biosystems, Foster City, CA, USA) and the KAPA Library Quantification Kit (Kapa Biosystems, Wilmington, MA, USA). Paired-end sequencing (2 × 300 bp) was performed on a MiSeq (Illumina, San Diego, CA, USA). The sequences obtained were pre-processed using Prinseq software to remove reads smaller than 35 bp and low-score sequences (lower than Phred 30) (Schmieder and Edwards 2011). Sequence reads were assembled using the software MIRA (Chevreux et al. 2004). The contigs and singletons were used in subsequent analyzes. In accordance with Tschoeke et al. (2014), we conducted a second assembly from the contigs obtained with Mira using the software CAP3 (Huang and Madan 1999). The gene prediction and functional annotation were performed using the RAST (rapid annotation using subsystem technology) program (Overbeek et al. 2014). We used the three available complete genomes of Endozoicomonas, which are: E. elysicola DSM 22380T, E. montiporae LMG 24815T and E. numazuensis DSM 25634T (NCBI project accession numbers: NZ_AREW00000000, NZ_JOKG01000000, NZ_JOKH00000000, respectively) (Table S1).
16S rRNA analysis
The 16S rRNA gene sequences were obtained from GenBank (NCBI) and aligned by CLUSTALW (Larkin et al. 2007) alignment method. The phylogenetic analyses were conducted using MEGA 6 (Tamura et al. 2013). The phylogenetic inference was based on Neighbour-joining method (Nei 1987) using Kimura 2P+G as nucleotide substitution model which was estimated from the data. Distance estimations were obtained by the model of Jukes & Cantor (Jukes and Cantor 1969). The support branches of tree topology were checked by 1000 bootstrap replications.
Genomic microbial taxonomy
In silico DNA–DNA Hybridization or Genome-to-Genome Distance (GGD) (Auch et al. 2010), amino acid identity (AAI) and average nucleotide identity (ANI) were calculated as described previously (Thompson et al. 2013a), with intra-population genomic relatedness ranging from 95 to 100 % AAI and ANI. The genome distance was calculated using genome-to-genome distance calculator (Meier-Kolthoff et al. 2014) with intra-population genomic similarity ranging from 70 to 100 %.
Genome-based phenotype
For the phenotypic analysis based on genome sequences (Amaral et al. 2014), we used 11 diagnostic biochemical features that have been applied in previous studies to identify species of Endozoicomonas (Yang et al. 2010; Mendoza et al. 2013). For each diagnostic feature, we searched for the corresponding genes. If a gene (or genes) involved in a phenotype is present in the genome, the organism is considered positive for this phenotype. The genes coding for proteins involved in those features were detected using RAST program (Overbeek et al. 2014). Genes associated with related biochemical pathways were identified with BLASTP algorithm (Altschul et al. 1990). We performed antiSMASH 2.0 software pipeline (Blin et al. 2014) for the automated identification of secondary metabolite biosynthesis clusters in whole genome sequences of bacteria.
In vitro phenotypic and chemotaxonomic characterization
Phenotypical characterization of the novel isolates and of the type strain of their close phylogenetic neighbour was performed using the commercial kit API 20E (bioMérieux) and Vitek 2 system (bioMérieux), following the manufacturer’s instructions. Tolerance to various NaCl concentrations (0.5 and 1.0–5.0 % {w/v} at increments of 1 %) and temperatures (4–37 °C) were tested on TSB and Marine Agar media. Growth was determined by measuring the turbidity (OD600) of cultures grown at various NaCl concentrations and temperatures. Motility test was conducted on semi-solid marine agar and it was determined by stab inoculation into tubes of classical formula of motility test medium. A positive test showed diffuse growth away from the stab line of inoculation, evidenced by turbidity, cloudiness, or feathery protuberances extending laterally throughout the medium. A negative motility was defined by growth confined to the stab line. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis was conducted for the novel isolates and for the type strain of their close phylogenetic neighbour, using the Vitek MS system (VITEK MS RUO; Shimadzu, Champs-sur-Marne, France) according to the manufacturer’s instructions. Briefly, a portion of a fresh colony was smeared onto a Vitek MS DS target slide and the preparations were overlaid with one microlitre of α-cyano-4-hydroxycinnamic acid (CHCA; bioMérieux) as matrix solution. After drying, the target plate was loaded into the Vitek MS mass spectrometer and air-dried for 1–2 min at room temperature. The system was calibrated externally with the mass spectrum obtained from fresh cells of the E. coli ATCC 8739 strain. The resulting peak lists were exported and analysed using SARAMIS software (bioMérieux VITEK MS RUO) for spectra comparison. Clusters were produced by hierarchical agglomerative clustering using the SARAMIS absolute or relative similarity measure and single-linkage criterion. Cluster analysis was performed using SARAMIS computing dendrograms based on similarities between masses.
Results and discussion
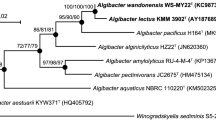
The phylogenetic analysis based on the 16S rRNA gene sequences showed that Endozoicomonas sp. strains CBAS 572T and CBAS 573 shared 99.5 % of identity (Fig. 1). The closest species of Endozoicomonas sp. strains CBAS 572T and CBAS 573 were E. numazuensis and E. montiporae, sharing >98 % of identity (Table 1). Bootstrap replicates strongly support the branches.

Phylogenetic tree of partial 16S rRNA gene sequences (1427 sites) based on neighbor-joining method and 1000 bootstrap replicates. Estimated nucleotide substitution model was Kimura 2P+G. Bootstrap values are shown. Hahella species were used as outgroup
Genome analysis
A total of 1,000,000 paired-end reads were generated for Endozoicomonas sp. strain CBAS 572T. The reads were assembled in 329 contigs. The coverage of the genome was 147-fold. The estimated genome size is 6,453,651 bp. The G+C content is 47.6 %. The number of coding sequences (CDS) is 5910. The number of RNA sequences is 138, of which 115 are tRNAs and 23 are rRNAs. For the Endozoicomonas sp. strain CBAS 573 a total of 1,819,409 paired-end reads were generated. The reads were assembled in 324 contigs. The coverage of the genome was 138-fold. The estimated size of the genome is 6,720,257 bp. The G+C content is 47.7 %. The number of coding sequences (CDS) is 6357. The number of RNA sequence is 135, of which 106 are tRNAs and 29 are rRNAs.
Genomic delineation of Endozoicomonas arenosclerae sp.nov.
AAI and ANI genomic analyses revealed less than 78 % identity values among Endozoicomonas strains CBAS 572T and CBAS 573 and their closest neighbours E. numazuensis and E. montiporae, whereas between strains CBAS 572T and CBAS 573 the identity values were 96.5 and 97 % (Table 1), respectively. GGD analysis found 99.2 % (±0.37) similarity between Endozoicomonas strains CBAS 572T and CBAS 573 and less than 27.50 % (±2.43) similarity among them and E. numazuensis and E. montiporae (Table 1). It is becoming clear that bacterial species can be defined on the basis of these features. A common definition consider that strains from the same species share at least 98.7 % 16S rRNA gene sequence similarity, >95 % of AAI and ANI, and >70 % In silico GGD (Thompson et al. 2014). These data support the conclusion that the isolates studied present a new species E. arenosclerae sp. nov.
In vitro phenotypic and chemotaxonomic features
In vitro phenotypic analysis were performed for CBAS 572T and CBAS 573 and for the close species E. montiporae LMG24815T. Cells grew at 12–35 °C and tolerated salinity from 2 to 5 %. Optimum growth occurs at 20–30 °C with 3 % NaCl. Unlike the neighbour species, it exhibited motility. Based on the analysis with API 20E, CBAS 572T and CBAS 573 utilize l-arginine and gelatin, but not 2-nitrophenyl-ß-d-galactopyranoside, l-lysine, l-ornithine, trisodium citrate, sodium thiosulfate, l-tryptophan (TDA), l-tryptophan (IND), sodium pyruvate, d-glucose, d-mannitol, inositol, d-sorbitol, l-rhamnose, d-sucrose, d-melibiose, amygdalin and l-arabinose, while E. montiporae LMG24815T does, except for sodium thiosulfate, urea, l-tryptophan (IND), inositol, l-rhamnose and l-arabinose. Vitek analysis showed that CBAS 572T had activity for phosphatase but not for ala-phe-pro-arylamidase, adonitol, l-pyrrolidonyl-arylamidase, l-arabitol, d-cellobiose, ß-galactosidase, H2S production, glutamyl arylamidase, d-glucose, gamma-glutamyl-transferase, glucose, ß–glucosidase, d-maltose, d-mannitol, d-mannose, ß-xylosidase, ß-alanine arylamidase, l-proline arylamidase, lipase, palatinose, tyrosine arylamidase, urease, d-sorbitol, saccharose, d-tagatose, trehalose, citrate (sodium), malonate, 5-keto-d-gluconate, l-lactate alkalinisation, alpha-glucosidase, succinate alkalinisation, ß-n-acetyl-galactosaminidase, α-galactosidase, glycine arylamidase, ornithine decarboxylase, lysine decarboxylase, l-histidine assimilation, coumarate, ß-glucuronidase, O-129 resistance, glu-gly-arg-arylamidase, l-malate assimilation, Ellman, l-lactate assimilation, while CBAS 573 was positive for phosphatase, ala-phe-pro-arylamidase, l-proline arylamidase and urease. CBAS 572T differs from E. montiporae LMG24815T by d-glucose, gamma-glutamyl-transferase, glucose, ß-glucosidase, d-maltose, d-mannitol, d-mannose, l-proline arylamidase, urease, d-sorbitol, saccharose-sucrose, trehalose, citrate (sodium), l-lactate alkalinisation, succinate alkalinisation, ß-n-acetyl-galactosaminidase, ornithine decarboxylase, lysine decarboxylase, coumarate, O-129 resistance and glu-gly-arg-arylamidase, which were positive for E. montiporae LMG24815T. The MALDI-TOF MS profiles allowed separation of the novel isolates CBAS 572T and CBAS 573 from the type strain of their near phylogenetic neighbour species E. montiporae LMG24815T. CBAS 572T and CBAS 573 share only 22 % of mass identity with E. montiporae LMG24815T (data not shown).
Genome-based phenotypic diagnostic features
The genes coding for key phenotypic markers, currently used to identify Endozoicomonas species, were analyzed in the genome of Endozoicomonas strains CBAS 572T and CBAS 573. The diagnostic phenotypes of the Endozoicomonas species, obtained from the literature, were compared with the predicted phenotypes obtained from whole genome sequences (Table 2). Some useful phenotypic features were found, including C8 esterase, N-acetyl-β-glucosaminidase, citric acid, uridine, siderophore and resorcinol that differentiate the novel isolates from the closest phylogenetic neighbours: E. numazuensis and E. montiporae.
The isolates CBAS 572T and CBAS 573 are representatives of a novel species of the genus Endozoicomonas, for which the name Endozoicomonas arenosclerae sp. nov. is proposed.
Formal description of Endozoicomonas arenosclerae sp. nov.
Endozoicomonas arenosclerae (a.re.no.scle’rae. N.L. gen. n. arenosclerae, of the sponge Arenosclera brasiliensis). Cells are Gram-negative, aerobic, motile, 0.5–1.0 µm in diameter after incubation for 48 h at 30 °C. Growth occurs at 12–35 °C in the presence of 2–4 % NaCl. Optimum growth occurs at 20–30 °C in the presence of 3 % NaCl. Colonies are cream coloured, circular and convex with entire margin on Marine Agar. In API 20E, can utilise l-arginine and gelatin, but do not 2-nitrophenyl-ß-d-galactopyranoside, l-lysine, l-ornithine, trisodium citrate, sodium thiosulfate, l-tryptophan (TDA), l-tryptophan (IND), sodium pyruvate, d-glucose, d-mannitol, inositol, d-sorbitol, l-rhamnose, d-sucrose, d-melibiose, amygdalin and l-arabinose. In Vitek analysis shows activity for phosphatase but not for adonitol, l-pyrrolidonyl-arylamidase, l-arabitol, d-cellobiose, ß-galactosidase, H2S production, glutamyl arylamidase, d-glucose, gamma-glutamyl-transferase, glucose, ß–glucosidase, d-maltose, d-mannitol, d-mannose, ß-xylosidase, ß-alanine arylamidase, lipase, palatinose, tyrosine arylamidase, d-sorbitol, saccharose-sucrose, d-tagatose, trehalose, citrate (sodium), malonate, 5-keto-d-gluconate, l-lactate alkalinisation, alpha-glucosidase, succinate alkalinisation, ß-n-acetyl-galactosaminidase, α-galactosidase, glycine arylamidase, ornithine decarboxylase, lysine decarboxylase, l-histidine assimilation, coumarate, beta-glucuronidase, O-129 resistance, glu-gly-arg-arylamidase, l-malate assimilation, Ellman, and l-lactate assimilation. In silico phenotypes predicted from genome sequences suggests strains are positive for alkaline phosphatase, n-acetyl-ß-glucosaminidase, citric acid, succinic acid, l-alanine, l-serine, thymidine, glycerol, bacteriocin, siderophore and resorcinol and negative for C8 esterase, l-fucose, uridine and ectoine. The type strain CBAS 572T (=Ab112T) has a DNA G+C content of 47.6 mol%.
Nucleotide sequence accession numbers
The Whole Genome Shotgun Projects for E. arenosclerae CBAS 572T (=Ab112T) and E. arenosclerae and CBAS 573 (=Ab227_MC) have been deposited in DDBJ/EMBL/GenBank under accession numbers LASA010000000 and LASB010000000, respectively.
References
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. doi:10.1016/S0022-2836(05)80360-2
Amaral GRS, Dias GM, Wellington-Oguri M, Chimetto L, Campeão ME, Thompson FL, Thompson CC (2014) Genotype to phenotype: identification of diagnostic vibrio phenotypes using whole genome sequences. Int J Syst Evol Microbiol 64:357–365. doi:10.1099/ijs.0.057927-0
Auch AF, von Jan M, Klenk H-P, Göker M (2010) Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2:117–134. doi:10.4056/sigs.531120
Blin K, Kazempour D, Wohlleben W, Weber T (2014) Improved lanthipeptide detection and prediction for antiSMASH. PLoS One. doi:10.1371/journal.pone.0089420
Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WE, Wetter T, Suhai S (2004) Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14:1147–1159. doi:10.1101/gr.1917404
Choi EJ, Kwon HC, Sohn YC, Yang HO (2010) Kistimonas asteriae gen. nov., sp. nov., a gammaproteobacterium isolated from Asterias amurensis. Int J Syst Evol Microbiol 60:938–943. doi:10.1099/ijs.0.014282-0
Du Z, Zhang W, Xia H, Lü G, Chen G (2010) Isolation and diversity analysis of heterotrophic bacteria associated with sea anemones. Acta Oceanol Sin 29:62–69. doi:10.1007/s13131-010-0023-1
Goffredi SK, Johnson SB, Vrijenhoek RC (2007) Genetic diversity and potential function of microbial symbionts associated with newly discovered species of Osedax polychaete worms. Appl Environ Microbiol 73:2314–2323. doi:10.1128/AEM.01986-06
Haley BJ, Grim CJ, Hasan NA et al (2010) Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholerae. BMC Microbiol 10:154. doi:10.1186/1471-2180-10-154
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9:868–877. doi:10.1101/gr.9.9.868
Hyun DW, Shin NR, Kim MS et al (2014) Endozoicomonas atrinae sp. nov., isolated from the intestine of a comb pen shell Atrina pectinata. Int J Syst Evol Microbiol 64:2312–2318. doi:10.1099/ijs.0.060780-0
Jukes TH, Cantor CR (1969) Evolution of protein molecules. Mamm Protein Metab. 3: 21–132. 10.1093/bioinformatics/btm404
Kurahashi M, Yokota A (2007) Endozoicomonas elysicola gen. nov., sp. nov., a c -proteobacterium isolated from the sea slug Elysia ornata. Syst Appl Microbiol 30:202–206. doi:10.1016/j.syapm.2006.07.003
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Martínez-García M, Díaz-Valdés M, Wanner G et al (2007) Microbial community associated with the colonial ascidian Cystodytes dellechiajei. Environ Microbiol 9:521–534. doi:10.1111/j.1462-2920.2006.01170.x
Meier-Kolthoff JP, Klenk HP, Göker M (2014) Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int J Syst Evol Microbiol 64:352–356. doi:10.1099/ijs.0.056994-0
Mendoza M, Güiza L, Martinez X et al (2013) A novel agent (Endozoicomonas elysicola) responsible for epitheliocystis in cobia Rachycentrum canadum larvae. Dis Aquat Organ 106:31–37. doi:10.3354/dao02636
Mohamed NM, Cicirelli EM, Kan J et al (2008) Diversity and quorum-sensing signal production of Proteobacteria associated with marine sponges. Environ Microbiol 10:75–86. doi:10.1111/j.1462-2920.2007.01431.x
Moreira AP, Duytschaever G, Tonon LAC et al (2014) Vibrio madracius sp. nov. Isolated from Madracis decactis (Scleractinia) in St Peter & St Paul Archipelago, Mid-Atlantic Ridge, Brazil. Curr Microbiol. doi: 10.1007/s00284-014-0600-1
Neave MJ, Michell CT, Apprill A, Voolstra CR (2014) Whole-genome sequences of three symbiotic Endozoicomonas Bacteria. Genome Announc 2:4–5. doi:10.1128/genomeA.00802-14.Copyright
Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Nishijima M, Adachi K, Katsuta A, Shizuri Y, Yamasato K (2013) Endozoicomonas numazuensis sp. nov., a gammaproteobacterium isolated from marine sponges, and emended description of the genus Endozoicomonas Kurahashi and Yokota 2007. Int J Syst Evol Microbiol 63: 709–714. doi: 10.1099/ijs.0.042077-0
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R (2014) The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucl Acids Res. doi:10.1093/nar/gkt1226
Pike RE, Haltli B, Kerr RG (2013) Description of Endozoicomonas euniceicola sp. nov. and Endozoicomonas gorgoniicola sp. nov., bacteria isolated from the octocorals Eunicea fusca and Plexaura sp., and an emended description of the genus Endozoicomonas. Int J Syst Evol Microbiol 63:4294–4302. doi:10.1099/ijs.0.051490-0
Pitcher DG, Saunders N, Owen RJ (1989) Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett Appl Microbiol 8:151–156. doi:10.1111/j.1472-765X.1989.tb00262.x
Rua CPJ, Trindade-Silva AE, Appolinario LR, Venas TM, Garcia GD, Carvalho LS, Lima A, Kruger R, Pereira RC, Berlinck RGS, Valle RAB, Thompson CC, Thompson F (2014) Diversity and antimicrobial potential of culturable heterotrophic bacteria associated with the endemic marine sponge Arenosclera brasiliensis. PeerJ 2:e419. doi:10.7717/peerj.419
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. doi:10.1093/bioinformatics/btr026
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30: 2725–2729. doi: 10.1093/molbev/mst197
Thompson CC, Vicente ACP, Souza RC, Vasconcelos ATR, Vesth T, Alves N, Ussery DW, Iida T, Thompson FL (2009) Genomic taxonomy of vibrios. BMC Evol Biol 9:258. doi:10.1186/1471-2148-9-258
Thompson CC, Vieira NM, Vicente A, Thompson F (2011a) Towards a genome based taxonomy of mycoplasmas. Infect Genet Evol 11:1798–1804. doi:10.1016/j.meegid.2011.07.020
Thompson FL, Thompson CC, Dias GM, Naka H, Dubay C, Crosa JH (2011b) The genus Listonella macdonell and colwell 1986 is a later heterotypic synonym of the genus Vibrio pacini 1854 (approved lists 1980)-a taxonomic opinion. Int J Syst Evol Microbiol 61:3023–3027. doi:10.1099/ijs.0.030015-0
Thompson CC, Chimetto L, Edwards RA, Swings J, Stackebrandt E, Thompson FL (2013a) Microbial genomic taxonomy. BMC Genom 14:913. doi:10.1186/1471-2164-14-913
Thompson CC, Silva GZ, Vieira NM, Edwards R, Vicente AC, Thompson FL (2013b) Genomic taxonomy of the genus Prochlorococcus. Microb Ecol 66:752–762. doi:10.1007/s00248-013-0270-8
Thompson CC, Emmel VE, Fonseca EL, Marin MA, Vicente ACP (2013c) Streptococcal taxonomy based on genome sequence analyses. F1000Research 2:67. doi:10.12688/f1000research.2-67.v1
Thompson CC, Amaral GR, Campeão M, Edwards RA, Polz MF, Dutilh BE, Ussery DW, Sawabe T, Swings J, Thompson FL (2014) Microbial taxonomy in the post-genomic era: rebuilding from scratch? Arch Microbiol. doi:10.1007/s00203-014-1071-2
Tschoeke DA, Moreira AP, Chimetto Tonon LA, de Mesquita MM, Gregoracci GB, Gomez-Gil B, Valle R, Thompson CC, Thompson FL (2014) Exploring the genome of cheese starter lactic acid bacterium Lactococcus lactis subsp. lactis CECT 4433. Genome Announc. doi:10.1128/genomeA.01142-14
Yang C, Chen M, Arun AB, Chen CA, Wang JT, Chen WM (2010) Endozoicomonas montiporae sp. nov., isolated from the encrusting pore coral Montipora aequituberculata. Int J Syst Evol Microbiol. doi: 10.1099/ijs.0.014357-0
Zielinski FU, Pernthaler A, Duperron S, Raggi L, Giere O, Borowki C, Dubilier N (2009) Widespread occurrence of an intranuclear bacterial parasite in vent and seep bathymodiolin mussels. Environ Microbiol 11:1150–1167. doi:10.1111/j.1462-2920.2008.01847.x
Acknowledgments
The authors thankfully acknowledge CNPq, CAPES, and FAPERJ for their founding and support for this project.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Appolinario, L.R., Tschoeke, D.A., Rua, C.P.J. et al. Description of Endozoicomonas arenosclerae sp. nov. using a genomic taxonomy approach. Antonie van Leeuwenhoek 109, 431–438 (2016). https://doi.org/10.1007/s10482-016-0649-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-016-0649-x