
Abstract
This review provides an overview of new technologies for DNA manipulations in actinomycetes exploiting recombinogenic engineering (Flp-FRT, Cre-loxP, Dre-rox, Tn5, GusA and I-SceI systems). We will describe some new vectors recently developed for engineering of complex phenotypes in actinomycetes. Several site-specific recombinases, transposons, reporter genes and I-SceI endonuclease have been utilized for genome manipulation in actinomycetes. Novel molecular tools will help to overcome many technical difficulties and will encourage new efforts to address the function of actinomycete genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent whole genome sequencing programs have revealed that the biosynthetic potential of Actinomycetales has been greatly underestimated. A common feature of all actinomycetes genomes is a length of about 8–10 Mb and the presence of approximately 20–30 gene clusters encoding the synthesis of secondary metabolites (Bentley et al. 2002; Ikeda et al. 2003; Oliynyk et al. 2007). In fact, the number of genes encoding biosynthetic enzymes in various actinomycetes clearly outnumbers the known secondary metabolites of these organisms. A reason for this observation might be that only a subset of biosynthetic pathway genes is expressed under standard laboratory culture conditions and therefore only a minority of potential chemical structures is produced. Such silent genetic loci are referred to as “cryptic” or “orphan” pathways. To exploit the natural biogenetic capability of microorganisms the biosynthesis of these putative metabolites needs to be induced. With the advent of next-generation DNA sequencing techniques, we gain access to a huge amount of genetic information, which need to be turned into new chemical and biological entities. Therefore, novel and efficient molecular genetic tools, which help to characterize gene functions and exploit the huge potential of actinomycetes are highly desirable. Here we describe novel genome manipulation techniques based on site-specific recombinases, transposon mutagenesis, rare-cutting meganucleases and gusA reporter system, developed for actinomycetes during the last 5 years.
Site-specific recombinases
Site-specific recombination (SSR) systems became powerful and efficient tools for high-throughput genetic analysis in the post genomic era (Branda and Dymecki 2004). Site-specific genome recombination systems have been described for a number of bacteria and higher organisms (Kuhn and Torres 2002; Leibig et al. 2008; Marx and Lidstrom 2002). Typically, site-specific recombinases are applied for the construction of unmarked mutants (Suzuki et al. 2005), targeting of heterologous DNA into the chromosome (Kuhstoss et al. 1991), marker-free expression of foreign genes (Schweizer 2003), transient and timed expression of genes (Slauch and Camilli 2000), deletion of large genomic fragments (Suzuki et al. 2005), and in vivo cloning of genomic DNA regions among others (Schweizer 2003). Most SSR systems are based on Cre/loxP from the P1 phage and Flp/FRT from yeast (Schweizer 2003; Branda and Dymecki 2004). The Cre and Flp proteins are bidirectional tyrosine recombinases catalysing reciprocal SSR of DNA at 34 bp target sites (loxP and FRT, respectively), which results in either excision or inversion depending on whether the target sites are located as direct or inverted repeats. Both recombinases do not require any host cofactors or accessory proteins, therefore could be functional in most living organisms (Kuhn and Torres 2002). Both recombinases have been successfully expressed in actinomycetes. In the next subchapters, we will present several strategies for genome engineering employing different SSR systems.
Construction of unmarked mutants
Study of the phenotypic changes in gene disruption mutants is a widely used approach to characterise gene function. The generation of a gene knockout relies upon the homologous recombination based methodology, which is usually an inefficient process in actinomycetes. Therefore, methods adapting a redET based technology to streptomycetes has attracted a lot of attention. Gust and colleagues have used λ-Red to promote recombination in Escherichia coli between a PCR-amplified antibiotic resistance cassette and Streptomyces coelicolor DNA on a cosmid from the S. coelicolor genome. The amplified cassette includes an origin of transfer (oriT; RK2) allowing the conjugal transfer of the PCR-targeted cosmid into S. coelicolor with a high frequency (Gust et al. 2003). Due to the large homologous fragments flanking the disrupting cassette, recombination process between the cosmid and the chromosome is more efficient. However, this strategy (as well as the others) most often results in the introduction of a selectable marker into the genome, which remains in the chromosome permanently (Kieser et al. 2000). The number of available resistance markers for actinomycete genome manipulations is limited and therefore the generation of multiple gene deletions in the same genetic background is also restricted. Resistance markers integrated into the chromosome may result in polar effects on the expression of genes located upstream and downstream. In addition, the expression of selectable markers may also influence bacterial fitness. These obstacles could be overcome if the resistance markers are eliminated after mutant selection, which can be achieved via SSR. Therefore, removal of a selectable marker from the genome using SSR has been frequently used in plants, mouse cell lines, yeast and recently in actinomycetes (Branda and Dymecki 2004; Schweizer 2003; Fedoryshyn et al. 2008a; Zelyas et al. 2009). SSR mediated excision of the antibiotic resistance cassette leaves a “scar” in the chromosome, meaning a single target site (e. g. loxP site in the case of Cre). The length of this scar is usually less than 50 bp and does not exert polar transcriptional effects. Marker rescue and reuse offers a simple and efficient way to introduce multiple gene deletions. However, consecutive deletions of additional genes in the same genetic background may be limited because of undesired DNA rearrangements generated by the multiple target sites left after each excision. Therefore, different recombinases should be used for the generation of multiple gene deletions. Several methods have been developed to produce unmarked mutants in microorganisms (Malaga et al. 2003; Siegl et al. 2010; Gust et al. 2003). To generate multiple mutations in actinomycetes, the recombinase based in-frame deletions are first constructed in E. coli and then introduced into the actinomycete via homologous recombination. This method was successfully applied to obtain S. coelicolor carrying eight separate deletions of the chaplin genes (Capstick et al. 2007). The major drawback of this approach is the absence of an antibiotic resistance marker for the double-crossingover selection. Therefore, large number of clones need to be screened to find the desired mutant.
Cre recombinase
In 2006, successful expression of the Cre recombinase in S. coelicolor A(3)2 was reported (Khodakaramian et al. 2006). The frequency of the resistance marker excision was from 60 to 90 %. However, this system did not become widely used, probably because the cre gene was introduced into streptomycetes on the φC31 phage, which is not a well distributed method of gene expression in streptomycetes. We have subsequently reported the successful expression of the synthetic gene encoding the Cre recombinase (Fedoryshyn et al. 2008b). This cre(a) gene can be expressed from different plasmids pALCRE, pNLCRE, pUWLHCRE and pUWLCRE (Table 1). The first two plasmids contain the temperature sensitive pSG5 replicon which is readily lost at temperatures above 37 °C. If plasmid replication is not supported from pSG5, pUWL-based plasmids carrying cre(a) may be used as well. They are usually lost after 2–3 generations of the actinomycetes culture without selective pressure. We have shown cre(a) to be successfully expressed in members of the Streptomyces, Micromonospora, Kitasatospora and Saccharothrix genera (Herrmann et al. 2012). The frequency of resistance marker excision by Cre expressed from pALCRE, pNLCRE, pUWLHCRE and pUWLCRE is nearly 100 % in all strains we have tested. To minimise genetic instability when performing multiple manipulations, different heterotypic lox sites containing mutations within the inverted repeats (loxLE and loxRE) have been used for eukaryotes and bacteria (Branda and Dymecki 2004; Leibig et al. 2008). Recombination of loxLE and loxRE results in a double mutant loxLERE site, which was reported to be a poor substrate for Cre (Leibig et al. 2008). We could show, however, that Cre recognizes the double mutated site at very high frequency in actinomycetes. Thus, the risk of undesired chromosomal rearrangements is still very high if Cre is iteratively used in combination with loxLE and loxRE (Herrmann et al. 2012).
Flp recombinase
The functional expression of the synthetic gene flp(a) encoding for Flp recombinase was reported in several actinomycetes: S. coelicolor M145, S. lividans TK24, and Saccharothrix espanaensis, but not in Micromonospora Tü6368 (Fedoryshyn et al. 2008b; Herrmann et al. 2012). However, the frequency of excision of a FRT flanked apramycin resistance gene from the chromosome of actinomycetes ranged from 10 to 40 %. This is significantly lower in comparison to Cre. The native flp gene has been expressed as well, showing an even lower marker excision frequency of around 2 % (Zelyas et al. 2009). However, even at this frequency it is possible to obtain marker free mutants relatively easy. The synthetic gene flp(a) is expressed from the pALFLP, pUWLHFLP and pUWLFLP plasmids (Table 1). Resistance markers flanked by FRT sites may be amplified from pIJ773 (an apramycin resistance gene) and from pIJ10700 (a hygromycin resistance gene) (Gust et al. 2003).
Dre recombinase
Recently, we have reported the very efficient expression of Dre recombinase in actinomycetes. Dre recombinase recognises a target site called rox, but not the FRT and loxP sites. The frequency of the marker removal from chromosomes of different actinomycetes was nearly 100 %. Thus, Dre recombinase is comparable with Cre in its high efficiency. Both recombinases do not show any cross-recognition of their target sites and therefore can be used to create marker free mutants in the same genetic background. As well as Cre, Dre recombinase was successfully expressed in Saccharothrix, Streptomyces, and Micromonospora genera (Herrmann et al. 2012). Dre recognises rox sites, which differ notably from the previously reported FRT and loxP sites in actinomycetes. This characteristic makes Dre an ideal partner for Flp and Cre in performing multiple mutations in a single genetic background.
Int and xis system of pSAM2
In the group of Prof. Pernodet, the xis and int genes from the pSAM2 plasmid have been used to delete antibiotic resistance markers from the chromosomes of streptomycetes. The authors generated several cassettes that contain antibiotic resistance genes inserted between the attL and attR sites (attL-antibiotic resistance-attR) and several vectors expressing the Int and Xis proteins from pSAM2 (Raynal et al. 2006). The successful functional expression of four different recombinases (Cre, Flp. Dre, and Xis/Int) in actinomycetes increases our ability to construct marker-free multi-mutant strains significantly. Theoretically, at least four consecutive resistance marker deletions can be performed in actinomycetes in a single genomic background.
Generation of the marker-free expression system
The stable integration of desired genetic elements into the chromosome has been exploited in many Streptomyces genome engineering strategies. The integration of regulatory genes and complementation of gene deletions is generally achieved using plasmids carrying a phage integrase gene (int) and the corresponding phage attachment site (attP). The most widely used integration vectors are based upon the Streptomyces phage φC31 and integrate within a chromosome condensation protein encoding gene. The other systems, based on the φBT1 and VWB phages, have gained popularity as well in the last years. The necessity to use an antibiotic selection and thus to leave behind a selection marker as well as additional unnecessary delivery vehicle sequences, such as an int gene and an E. coli origin of replication in integrative vectors creates the major limitations for using them. In addition, a constant expression of the int gene might also lead to an unstable construct. Therefore, two integrative vectors (pTES and pTOS, Fig. 2, Table 1) were generated, which allow the removal of unwanted delivery vehicle DNA like the phage integrase gene and the antibiotic selection marker after the successful integration of the genes of interest into the chromosome. pTES is φC31- based and its bacterial attachment site (attB) in the chromosome is located in a gene encoding a chromosome condensation protein, while the VWB-based vector pTOS integrates into a gene encoding the arginine tRNA. An excision of the φC31 integrase gene together with an apramycin resistance marker from the pTES vector is performed by Cre recombinase, while Dre recombinase removes the VWB integrase gene together with the apramycin resistance marker from the pTOS plasmid (Fig. 1). Both, pTES and pTOS vectors, enable the engineering of stable recombinant actinomycete strains devoid of any selection marker. The availability of the marker-free expression system allows the generation of multiple, stable, unmarked integrations and the expression of several genes simultaneously (Herrmann et al. 2012).

Schematic diagram depicting the mechanism of marker-free integration of a gene of interest (goi) into the streptomycetes genome. a The integrase gene on the plasmid is under the control of the native phage promoter and its product catalyses the integration of the plasmid between the attachment site on the plasmid (attP) and the attachment site on the bacterial chromosome (attB). b Selection for the integration of the plasmid into the genome is provided by the selection marker. c Expression of Dre recombinase will result in the deletion of the plasmid sequence between the rox-sites, thus removing the marker and leaving behind only the gene of interest and one rox-site in the genome. d Dre and Cre recognition sequences

Restriction maps of the vectors used for cloning of recombinases, I-SceI meganuclease, transposase and gusA. aac(3′)IV apramycin resistance conferring gene, aadA spectinomycin resistance conferring gene, attP attachment site on plasmid for phage integration, bla carbenicillin/ampicillin resistance conferring gene, ColE1ori origin of replication in E. coli, ermEp constitutive promoter in streptomycetes, hph hygromycin resistance conferring gene, loxP recognition site for Cre recombinase, int phage integrase gene, intp integrase promoter, I-SceI recognition site for I-SceI meganuclease, LacZa beta-galactosidase gene for blue/white selection, ori origin of replication in streptomycetes, oripUC18 origin of replication in E. coli, oriT origin of transfer, pSG5rep temperature sensitive replicon in streptomycetes, rox recognition site for Dre recombinase, T4 phage terminator sequence, tfd phage terminator sequence, tipAp thiostrepton-inducible promoter, tsr thiostreptone resistance conferring gene
Generation of large-scale deletions
The ability to delete and reintegrate large genomic fragments within actinomycete genomes is of great interest, especially for the investigation of antibiotic biosynthetic genes, which are usually clustered.
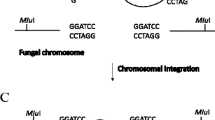
The strategy for the generation of gene cluster deletions based on the Cre/loxP system is presented in Fig. 3. Two loxP sites should be introduced on both ends of a gene cluster as direct repeats for the subsequent recombination. The specific advantage of the presented strategy is the integration of the first loxP site via a single-crossover, which are usually easier to obtain than double-crossover events. The usefulness of this method has been demonstrated recently by generating three deletions spanning the α-lipomycin, phenalinolactone and monensin biosynthetic gene clusters in S. aureofaciens Tü117, S. sp. Tü6071 and S. cinnamonensis A519, respectively. The successful complementation of phenalinolactone production by introducing the corresponding gene cluster opens many opportunities to modify a cosmid directly in E. coli via recombineering techniques and perform fast gene deletion using mutated cosmids (Herrmann et al. 2012).

Schematic diagram depicting the steps for deleting large gene clusters from the streptomycetes chromosome. Initially, a suicide plasmid carrying two recognition sites (rs) flanking a region homologous to the 5′ end of the cluster is introduced into the chromosome by a single-crossover. Expression of the site-specific recombinase leads to excision of the plasmid backbone, including the selection marker, and leaves one rs at the 5′ end of the cluster. A second suicide plasmid carrying one rs and a region homologous to the 3′ end of the cluster is integrated into the chromosome by means of single-crossover. Expression of the site-specific recombinase finally leads to excision of the entire cluster and loss of the resistance marker, leaving in its place only one rs “scar” in the chromosome
In addition to a systematic functional analysis of biosynthetic gene clusters, the Cre-mediated large fragments excisions have been utilised to construct the improved S. avermitillis host strains for the heterologous production of natural products. Strikingly, Cre recombinase was able to excise a fragment of more than 1.5 Mb in one step (Komatsu et al. 2010). Another way to create a “superhost” strain of S. coelicolor by deleting endogenous gene cluster (~170 kb DNA in total) has been described by Gomez-Escribano and Bibb 2010. They have used a homologous recombination to generate four large deletions on the chromosome. However, this approach seems to be more labour intensive and time consuming in comparison to the method involving site-specific recombinases.
In summary, the variety of different genome engineering strategies including the construction of unmarked multiple mutations, marker-free expression of target genes, large-fragment deletions, and chromosomal integration of biosynthetic gene clusters have been performed in actinomycetes using the site-specific recombinases Cre, Dre, Flp, and In/Xis of pSAM2. Obviously, these techniques will find a broad application in actinomycete genetics and will facilitate functional studies of a large number of genes from the increasing set of sequenced genomes.
In vivo transposon mutagenesis
Random mutagenesis tools like transposons have a long standing successful history in genetic manipulation of bacteria and higher organisms. Noteworthy is their huge impact on studying gene functions and generating recombinant strains (Goryshin et al. 2000). There have been several attempts to establish transposon mutagenesis in actinomycetes, including Tn5 derivatives, natural Streptomyces transposons and different IS-elements (Table 2) (Baltz et al. 1997; Gehring et al. 2000; Guenes et al. 1999; Ikeda et al. 1993; Solenberg and Baltz 1991; Volff and Altenbuchner 1997). One of the first, Tn4556 from S. fradiae, has been developed as a tool for random mutagenesis. Integration of this transposon and its derivatives, however, is not completely random (Ikeda et al. 1993). Moreover, it was recently observed that a derivative of Tn4556 causes a genetic instability near the native insertion sequence IS1649 in S. coelicolor (Wildenbrant and Kao 2007). The second native transposon, IS493 from S. lividans, appeares to be much less efficient than Tn4560 and has a strong insertion bias into the DNA regions with low G/C content (Solenberg and Baltz 1991). Baltz and colleagues have developed many transposons derived from IS493 and show their usefulness in a couple of streptomycetes. Transposon mutagenesis based on IS elements from related organisms may interfere with the indigenous transposons, or transpose in response to the introduction of related elements leading to unmarked mutations. Tn5493, a derivative of Tn5, was applied for transposon mutagenesis in S. lividans (Volff and Altenbuchner 1997). This is the only transposon from an unrelated organism, which has been applied to streptomycetes in vivo. Despite success in S. lividans TK64 (Volff and Altenbuchner 1997), Tn5493 was not used in other streptomycetes, likely because of its low efficiency in other actinomycetes. An improved version of Tn5 based transposon mutagenesis in streptomycetes was generated using optimised Tn5 mosaic end sequences (ME) and the GC-rich synthetic tnp(a) gene encoding a hyperactive transposase, which binds more efficiently to the transposon ends (Steiniger et al. 2006). The frequency of insertion for the hyperactive Tn5 derivative was 98 % of transformed cells, which is significantly higher than reported to date for in vivo random mutagenesis systems in streptomycetes (Baltz et al. 1997; Volff and Altenbuchner 1997). This transposon also contains an apramycin resistance marker, which is very efficient in most streptomycetes, and the R6Kγ replication origin, allowing quick and easy cloning of the insertion site directly from the chromosome (Fig. 4). The Tn5 transposon mutagenesis system, based on the hyperactive transposase and ME sequences was successfully applied to several streptomycetes strains: S. coelicolor, S. lividans. S. albus, S. fradiae Tü2717, S. globisporus 1912 and others (Petzke and Luzhetskyy 2009). Recently, a new mini-transposon encoding the apramycin resistance gene aac(3′)IV was generated based on IS204 from Nocardia asteroides YP21. The mini-transposon contains also an origin of replication for E. coli and therefore the insertion can be efficiently identified in the genome via rescued plasmid sequencing (Zhang et al. 2012). Available transposon systems were usually applied to S. coelicolor and S. lividans, therefore their utility in other important actinomycetes still needs to be proven. The features of some transposons are summarized in the Table 2.

Scheme depicting the steps of the transposon insertion site identification
Recently, an excellent high-throughput in vitro transposon mediated mutagenesis system of the S. coelicolor cosmid library has been developed by the group of Prof. Dyson (Fernández-Martínez et al. 2011). The Tn5 hypertransposase has been used for in vitro transposon mutagenesis of the ordered S. coelicolor cosmid library followed by conjugal intergeneric transfer of a mutated cosmid to S. coelicolor and selection for allelic replacement. 311 cosmids were mutated using the in vitro Tn5 transposon-tagging technology resulting in a library with 6,482 disrupted genes. Although this system is very useful to mutagenise the S. coelicolor genome, its use in other strains is limited by the commercial availability of a cosmid library.
I-SceI endonuclease
The rare-cutting homing endonucleases have been recognized as efficient tools to facilitate targeted mutagenesis in many organisms e.g. Bacillus anthracis, E. coli, Burkhodelia and others (Posfai et al. 1999; Lopez et al. 2009; Janes and Stibiz 2006) These enzymes introduce a double strand break into the DNA and thus stimulate homologous recombination at a certain locus. The endonuclease I-SceI has been used in streptomycetes as a chromosomal double strand break inducing agent (Jasin 1996). Using this system it was shown that homologous recombination is the major pathway involved in DNA double strand break repair in streptomycetes. Theoretically, an 18 bp recognition sequence appears once in 70 billion bases of a random sequence. Genomes of streptomycetes analyzed to date are free of I-SceI sites, abolishing the risk of genome fractionation by the meganuclease. Considering this fact the I-SceI based system was used to facilitate gene targeting. A step by step procedure is presented in Fig. 5. The mutant allele is cloned into the suicide plasmid which contains an antibiotic selection marker and a recognition site for I-SceI. This plasmid is introduced into the streptomycete and integrates into the chromosome via homologous recombination. Then, the I-SceI endonuclease is expressed in the resulting mutant under the inducible promoter tipA from pALSCE or under the constitutive promoter ermEp from pUWLHSCE. Both pALSCE and pUWLHSCE are replicative vectors, which can be lost again from the mutant under non-seletive conditions. In addition, pALSCE carries a temperature sensitive replicon for streptomycetes and is readily lost after incubation above 37 °C for 2 days. The double strand break introduced by I-SceI stimulates intramolecular recombination between parental and mutant alleles. Therefore, 25 times fewer colonies have to be screened in order to find a mutant than compared to the approach without I-SceI. Using allele-specific primers, Streptomyces cells carrying the mutated gene can easily be identified by PCR analysis. An additional advantage of the method is that I-SceI serves not only as a stimulator of homologous recombination at the target locus, but also as a counter-selection tool against cointegrates. The marker-free nature of the mutations facilitates the creation of multiple gene deletions in a single genomic background. In addition, this molecular tool can be applied to study DNA double strand break repair mechanisms in streptomycetes (Siegl et al. 2010).

Example of the I-SceI-based targeted gene inactivation in streptomycetes. Homologous recombination between the mutant and the wild-type alleles leads to a cointegrate. Cleavage of the chromosome by I-SceI stimulates recombination between homologous regions resulting either in a reversion to the wild type or in a marker-less gene replacement event
GusA based reporter system
Reporter genes have found numerous applications in bacterial genetic systems. They are used to study gene regulation, gene expression activity, translation and transcription of operons, gene targeting improvement and many others. The current set of reporter systems available for streptomycetes includes neo and cat resistance genes (Ward et al. 1986; Shaw and Hopwood 1976), catechol 2,3-dioxygenase (Zukowski et al. 1983; Sekurova et al. 2004), green fluorescent protein (Sun et al. 1999), luciferase (Craney et al. 2007) and since recently gusA (Myronovskyi et al. 2012). The most widely used reporters in actinomycetes are GFP and luciferase. While GFP is a powerful tool to monitor the trafficking and subcellular localization of proteins, its use for gene expression studies is limited due to low sensitivity (no enzymatic signal amplification) (Hutter 2006). In addition, the assay may suffer from UV-induced toxicity and photobleaching, thus limiting the duration of observation and analysis. In several systems, the formation of GFP aggregates causes cytotoxicity (Haseloff and Amos 1995; Crameri et al. 1996). Moreover, actinomycetes often have a high level of autofluorescence, which makes the analysis more complicated because of a low signal-to-noise ratio (Kieser 2000). Luciferase assays can be rapid and sensitive. The short half-life of luciferases makes them convenient for monitoring the dynamics of gene transcription (Nordeen 1988). The lack of autoluminescence in actinomycetes results in a high signal-to-noise ratio when using luciferases (Craney et al. 2007). The disadvantage is the complexity of enzymatic reactions, which depend on multiple reagents. For example, bacterial luciferase uses FMNH2, O2, and a ten-carbon aldehyde, decanal, whereas eukaryotic luciferases use ATP, Mg2 + O2, and the substrate luciferin. In addition, the luciferases do not enzymatically amplify the signal, they are labile, and have a short half-life (Wilson and Hastings 1998).
The gusA based reporter system was recognized because of a number of advantages: extreme sensitivity, simple assays, enzyme stability and the possibility of visual, spectrophotometric and fluorogenic assays. Many of its substrates, including X-gluc, MUG and p-nitrophenyl-β-D-glucuronide, are commercially available, which is critical for any enzymatic reporter system (Jefferson et al. 1987; Jefferson 1989). Most of the described actinomycete strains lack the indigenous β-glucuronidase activity. Several strategies of functional gene studies have been tested using the gusA based system and will be discussed in detail in the following sections.
Gene targeting
One of the applications for which the gusA reporter was used is the generation of gene inactivation vectors for rapid visual screening of clones with double-crossovers. For this purpose the gusA gene was introduced into the backbone of the common vectors pKC1132 and pKGLP2 (Fig. 2), suicidal vectors, and pKC1139, a replicative vector. Such an approach facilitates the selection of clones with double-crossovers by visual screening without picking thousands of colonies. Clones containing the vector will turn blue after overlaying substrate solution, and only clones that have lost the plasmid will stay white (Myronovskyi et al. 2012). The combination of the gusA gene and an antibiotic resistance marker for gene inactivation guarantees that nearly 100 % of the white clones contain the inactivated gene (Fig. 6). Successful utilization of gusA-containing vectors for gene inactivation indicates that this approach can significantly simplify the detection of gene inactivation even in problem strains with low frequencies of conjugation and crossover events. The gusA based gene inactivation system has been successfully tested in different actinomycete genera: Streptomyces, Kitasatospora and Saccharothrix.

Schematic diagram depicting the steps of targeted gene inactivation with the help of gusA
Transcriptional fusion
One of the most frequent uses of reporters is a transcriptional fusion. It is probably the easiest way to determine the expression level of any particular gene. Today the most commonly used streptomycetes reporters for transcriptional fusion are EGFP and luciferase. There are some reports on application of XylE. Utilization of EGFP is limited because a number of streptomycetes strains fluoresce in green light and specialized equipment is required to measure fluorescence (Willemse and van Wezel 2009). The main disadvantage of XylE is that the enzymatic assay is lethal to streptomycetes cells, so it is impossible to pick viable clones from plates after the assay (Ingram et al. 1989). In this context, the gusA gene has no such limitations: the plate-based assays are not lethal and for quantification a simple spectrophotometer is required. Because of the enzymatic amplification of the signal, the assay is very sensitive. To establish transcriptional fusions with the gusA gene, the pGUS promoter probe vector (Fig. 2) was constructed and successfully applied. It contains a terminator cassette to avoid any read-through from the promoters in the 5′ region of the plasmid DNA (Myronovskyi et al. 2012). The simplicity, sensitivity and flexibility of the gusA transcriptional fusion method demonstrate that it is a good alternative to the widely used EGFP and XylE fusions.
Translational fusion
Transcriptional fusions can be used only to monitor the mRNA level. For this type of fusion, the mRNA of gusA is produced, and all other post-transcriptional events are the same as for gusA mRNA. Thus, any post-transcriptional modifications of the expression of the gene of interest could not be monitored. A well-known example of transcription without translation in streptomycetes is bld regulation, during which certain genes are transcribed, but translation occurs only if the rare leucine TTA codon–decoding tRNA is present (Takano et al. 2003). Therefore, translational fusions that result in the production of a hybrid protein are of great importance. The GusA protein tolerates large C- and N-terminal fusions without loss of activity (Schmitz et al. 1990), so we constructed the vector pGUSHL4aadA for N-terminal fusions with gusA. In our system, GusA translational fusions produce a hybrid protein with amino-terminal sequences derived from a protein of interest and a carboxy-terminal fragment of GusA (Myronovskyi et al. 2012). In this case, any post-transcriptional effects on the expression of a gene of interest will be reflected in GusA production. The translational fusions with gusA can be used in actinomycetes to directly, quantitatively measure protein yields. Several possible applications of this system include screening for colonies with activated silent genes or pathways, studying the factors affecting translation efficiency, and selecting for the heterologous host yielding the largest amounts of target protein. All of these applications can be realized by semi-quantitative visual inspection directly on plates, as well as by quantitative spectrophotometric or fluorogenic measurement.
References
Baltz RH, McHenney MA, Cantwell CA, Queener SW, Solenberg PJ (1997) Applications of transposition mutagenesis in antibiotic producing streptomycetes. Antonie Van Leuwenhoek 71:179–187
Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O′Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147
Bhatt A, Kieser T (1999) Transposition of IS117 of Streptomyces coelicolor A3(2) in Mycobacterium smegmatis. Microbiology 145:1201–1207
Bierman M, Logan R, O’Brian K, Seno ET, Rao RN, Schoner BE (1992) Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49
Branda CS, Dymecki SM (2004) Talking about a revolution: the impact of site-specific recombinases on genetic analyses in mice. Dev Cell 6:7–28
Capstick DS, Willey JM, Buttner MJ, Elliot MA (2007) SapB and the chaplins: connections between morphogenetic proteins in Streptomyces coelicolor. Mol Microbiol 64:602–613
Crameri A, Whitehorn E, Tate E, Stemmer W (1996) Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol 14:315–319
Craney A, Hohenauer T, Xu Y, Navani NK, Li Y, Nodwell J (2007) A synthetic luxCDABE gene cluster optimized for expression in high-GC bacteria. Nucl Acids Res 35:e46–e46
Fedoryshyn M, Petzke L, Welle E, Bechthold A, Luzhetskyy A (2008a) Marker removal from actinomycetes genome using Flp recombinase. Gene 419:43–47
Fedoryshyn M, Welle E, Bechthold A, Luzhetskyy A (2008b) Functional expression of the Cre recombinase in actinomycetes. Appl Microbiol Biotechnol 78:1065–1070
Fernández-Martínez LT, Del Sol R, Evans RM, Fielding S, Herron PR, Chandra G, Dyson PJ (2011) A transposon insertion single-gene knockout library and new ordered cosmid library for the model organism Streptomyces coelicolor A3(2). Antonie Van Leeuwenhoek 99:515–522
Gehring AM, Nodwell JR, Beverley SM, Losick R (2000) Genomewide insertional mutagenesis in Streptomyces coelicolor reveals additional genes involved in morphological differentiation. Proc Natl Acad Sci USA 97:9642–9647
Gomez-Escribano JP, Bibb MJ (2011) Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb Biotechnol 4:207–215
Goryshin I, Jendrisak J, Hoffman L, Meis R, Reznikoff WS (2000) Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat Biotechnol 18:97–100
Guenes G, Smith B, Dyson P (1999) Genetic instability associated with insertion of IS6100 into one end of the Streptomyces lividans chromosome. Microbiology 145:2203–2208
Gust B, Challis GL, Fowler K, Kieser T, Chater KF (2003) PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci USA 100:1541–1546
Haseloff J, Amos B (1995) GFP in plants. Trends Genet 11:328–329
Herrmann S, Siegl T, Luzhetska M, Petzke L, Jilg C, Welle E, Erb A, Leadlay PF, Bechthold A, Luzhetskyy A (2012) Site-specific recombinase strategies for engineering actinomycetes genome. Appl Environ Microbiol 78:1804–1812
Hutter H (2006) Fluorescent reporter methods. Methods Mol Biol 351:155–173
Ikeda H, Takada Y, Pang CH, Tanaka H, Omura S (1993) Transposon mutagenesis by Tn4560 and applications with avermectin-producing Streptomyces avermitilis. J Bacteriol 175:2077–2082
Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol 21:526–531
Ingram C, Brawner M, Youngman P, Westpheling J (1989) xylE functions as an efficient reporter gene in Streptomyces spp.: use for the study of galP1, a catabolite-controlled promoter. J Bacteriol 171:6617–6624
Janes BK, Stibitz S (2006) Routine markerless gene replacement in Bacillus anthracis. Infect Immun 74:1949–1953
Jasin M (1996) Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet 12:224–228
Jefferson RA (1989) The GUS reporter gene system. Nature 342:837–838
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Khodakaramian G, Lissenden S, Gust B, Moir L, Hoskisson PA, Chater KF, Smith MC (2006) Expression of Cre recombinase during transient phage infection permits efficient marker removal in Streptomyces. Nucl Acids Res 34:e20
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. The John Innes Foundation, Norwich
Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H (2010) Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc Natl Acad Sci USA 107:2646–2651
Kuhn R, Torres RM (2002) Cre/loxP recombination system and gene targeting. Methods Mol Biol 180:175–204
Kuhstoss S, Richardson MA, Rao RN (1991) Plasmid cloning vectors that integrate site-specifically in Streptomyces spp. Gene 97:143–146
Leibig M, Krismer B, Kolb M, Friede A, Götz F, Bertram R (2008) Marker removal in Staphylococci via Cre recombinase and different lox sites. Appl Environ Microbiol 74:1316–1323
Lopez C, Rholl D, Trunk L, Schweizer P (2009) Versatile dual- technology system for markerless allele replacement in Burkholderia pseudomallei. Appl Environ Microbiol 20:6496–6503
Luzhetskyy A, Zhu L, Gibson M, Fedoryshyn M, Dürr C, Hofmann C, Hoffmeister D, Ostash B, Mattingly C, Adams V, Fedorenko V, Rohr J, Bechthold A (2005) Generation of Novel Landomycins M and O Through Targeted Gene Disruption. ChemBioChem 4:675–678
Malaga W, Perez E, Guilhot C (2003) Production of unmarked mutations in mycobacteria using site-specific recombination. FEMS Microbiol Lett 219:261–268
Marx CJ, Lidstrom ME (2002) Broad-host-range cre-lox system for antibiotic marker recycling in Gram-negative bacteria. Biotechniques 233:1062–1067
Myronovskyi M, Welle E, Fedorenko V, Luzhetskyy A (2011) Beta-glucuronidase as a sensitive and versatile reporter in actinomycetes. Appl Environ Microbiol 77:5370–5383
Nordeen SK (1988) Luciferase reporter gene vectors for analysis of promoters and enhancers. Biotechniques 6:454–458
Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25:447–453
Petzke L, Luzhetskyy A (2009) In vivo Tn5 transposon mutagenesis of streptomycetes. Appl Microbiol Biotechnol 83:979–986
Pitman A, Herron P, Dyson P (2002) Cointegrate resolution following transposistion of Tn1792 in Streptomyces avermitilis facilitates analysis of transposon-tagged genes. J Microbiol Methods 49:89–96
Posfai G, Kolisnychenko V, Bereczki Z, Blattner FR (1999) Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res 27:4409–4415
Raynal A, Karray F, Tuphile K, Darbon-Rongère E, Pernodet JL (2006) Excisable cassettes: new tools for functional analysis of Streptomyces genomes. Appl Environ Microbiol 72:4839–4844
Schmitz UK, Lonsdale DM, Jefferson RA (1990) Application of the beta-glucuronidase gene fusion system to Saccharomyces cerevisiae. Curr Genet 17:261–264
Schweizer HP (2003) Applications of the Saccharomyces cerevisiae Flp-FRT system in bacterial genetics. J Mol Microbiol Biotechnol 5:67–77
Sekurova ON, Brautaset T, Sletta H, Borgos SE, Jakobsen MO, Ellingsen TE, Strom AR, Valla S, Zotchev SB (2004) In vivo analysis of the regulatory genes in the nystatin biosynthetic gene cluster of Streptomyces noursei ATCC 11455 reveals their differential control over antibiotic biosynthesis. J Bacteriol 186:1345–1354
Shaw WV, Hopwood DA (1976) Chloramphenicol acetylation in Streptomyces. J Gen Microbiol 94:159–166
Siegl T, Petzke L, Welle E, Luzhetskyy A (2010) I-SceI endonuclease: a new tool for DNA repair studies and genetic manipulations in streptomycetes. Appl Microbiol Biotechnol 87:1525–1532
Slauch JM, Camilli A (2000) IVET and RIVET: use of gene fusions to identify bacterial virulence factors specifically induced in host tissues. Methods Enzymol 326:73–96
Solenberg PJ, Baltz RH (1991) Transposition of Tn5096 and other IS493 derivatives in Streptomyces griseofuscus. J Bacteriol 173:1096–1104
Steiniger M, Metzler J, Reznikoff WS (2006) Mutation of Tn5 transposase beta-loop residues affects all steps of Tn5 transposition: the role of conformational changes in Tn5 transposition. Biochemistry 45:15552–15562
Sun J, Kelemen GH, Fernández-Abalos JM, Bibb MJ (1999) Green fluorescent protein as a reporter for spatial and temporal gene expression in Streptomyces coelicolor A3(2). Microbiology 145:2221–2227
Suzuki N, Nonaka H, Tsuge Y, Inui M, Yukawa H (2005) New multiple-deletion method for the Corynebacterium glutamicum genome, using a mutant lox sequence. Appl Environ Microbiol 71:8472–8480
Takano E, Tao M, Long F, Bibb MJ, Wang L, Li W, Buttner MJ, Bibb MJ, Deng ZX, Chater KF (2003) A rare leucine codon in adpA is implicated in the morphological defect of bldA mutants of Streptomyces coelicolor. Mol Microbiol 50:475–486
Volff JN, Altenbuchner J (1997) High frequency transposition of the Tn5 derivative Tn5493 in Streptomyces lividans. Gene 194:81–86
Ward JM, Janssen GR, Kieser T, Bibb MJ, Buttner MJ, Bibb MJ (1986) Construction and characterisation of a series of multi-copy promoter-probe plasmid vectors for Streptomyces using the aminoglycoside phosphotransferase gene from Tn5 as indicator. Mol Gen Genet 203:468–478
Wildenbrant E, Kao C (2007) Introduction of the foreign transposon Tn4560 in Streptomyces coelicolor leads to genetic instability near the native insertion sequence IS1649. J Bacteriol 189:9108–9116
Willemse J, van Wezel GP (2009) Imaging of Streptomyces coelicolor A3(2) with reduced autofluorescence reveals a novel stage of FtsZ localization. PLoS ONE 4:e4242
Wilson T, Hastings JW (1998) Bioluminescence. Annu Rev Cell Dev Biol 14:197–230
Zelyas N, Tahlan K, Jensen SE (2009) Use of the native flp gene to generate in-frame unmarked mutations in Streptomyces ssp. Gene 443:48–54
Zhang X, Bao Y, Shi X, Ou X, Zhou P, Ding X (2012) Efficient transposition of IS204-derive plasmids in Streptomyces coelicolor. J Microbiol Methods 88:67–72
Zukowski MM, Gaffney DF, Speck D, Kauffmann M, Findeli A, Wisecup A, Lecocq JP (1983) Chromogenic identification of genetic regulatory signals in Bacillus subtilis based on expression of a cloned Pseudomonas gene. Proc Natl Acad Sci USA 80:1101–1105
Acknowledgments
The work in the laboratory of AL was supported by the BMBF (GenBioCom), DFG (Lu1524/2-1) and ERC (EXPLOGEN) grants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Siegl, T., Luzhetskyy, A. Actinomycetes genome engineering approaches. Antonie van Leeuwenhoek 102, 503–516 (2012). https://doi.org/10.1007/s10482-012-9795-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-012-9795-y