
Abstract
The aim of this study was to characterize and compare the bacterial community structure of two distinct oil samples from a petroleum field in Brazil by using both molecular, based on the construction of 16S rRNA gene libraries, and cultivation methods. Statistical comparisons of libraries based on Amplified Ribosomal DNA Restriction Analysis (ARDRA) data revealed no significant differences between the communities recovered in the non-biodegraded (NBD) and highly biodegraded oils (HBD). BlastN analysis of the 16S rRNA gene sequences representative of distinct ribotypes from both oils showed the presence of nine different bacterial genera in these samples, encompassing members of the genera Arcobacter, Halanaerobium, Marinobacter, Propionibacterium, Streptomyces, Leuconostoc, Acinetobacter, Bacillus and Streptococcus. Enrichments obtained using oil as inoculum and sole carbon source yielded bacterial isolates showing high 16S rRNA gene sequence similarity with Achromobacter xylosoxidans, Bacillus subtilis, Brevibacillus sp., Dietzia sp. and Methylobacterium sp. Comparison between the data obtained using cultivation-independent and enrichment cultures suggests that different selection of community members may occur when using distinct approaches. All the organisms found, except for Leuconostoc sp. and Streptococus sp., have been previously reported in the literature as hydrocarbon degraders and/or associated to oil field environments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biodegradation has been a great issue for oil exploration in many prolific basins worldwide due to a systematic decrease in paraffin content and an increase in oil density, sulfur content, acidity and viscosity (Connan 1984; Larter et al. 2003; Larter et al. 2006), with negative economic consequences for oil production and refining operations. Living organisms are able to survive in oil field environments if certain physical characteristics and chemical composition are favorable. Microorganisms inhabiting these environments may destroy hydrocarbons and other oil components yielding altered and undesirable “heavy oil” (Head et al. 2003). Although the in situ metabolic activities of petroleum microorganisms are poorly understood, a wide variety of bacterial and archaeal groups, including strictly anaerobic thermophilic and hyperthermophilic microorganisms as well as facultative anaerobic and thermotolerant ones, have been detected ex-situ or isolated from oil reservoirs (Magot et al. 2000).
Culture-based techniques have traditionally been the primary tools used for studying the microbiology of different environments (Chandler et al. 1997). These approaches, while extremely important for understanding the physiological potential of isolated organisms, do not necessarily provide comprehensive information on the composition of microbial communities (van Hamme et al. 2003). Reliable characterization of microbial communities became possible due to the introduction of molecular methods in microbial ecology studies, which have proven to be effective for evaluation of microbial communities in diverse environments such as ocean (DeLong 1992), soil and sediments (Takami et al. 1997) and hot springs (Hugenholtz et al. 1998). The recovery of DNA directly from environmental samples, with subsequent amplification by polymerase chain reaction (PCR), cloning and sequencing of 16S rRNA genes, has unraveled an astonishing microbial diversity in a variety of habitats (Pace 1996; Hugenholtz et al. 1998). In addition, the cultivation-independent approach allows the detection and phylogenetic identification of fastidious or as yet uncultured organisms (Juck et al. 2000). In petroleum microbiology, the use of molecular techniques in community analyses of oil fields is still recent and scarce (Voordouw et al. 1996; Orphan et al. 2000; Magot et al. 2000; Bonch-Osmolovskaya et al. 2003), despite their huge potential to help culture-based techniques to gather important and complementary sets of data.
The purpose of this research was to evaluate the bacterial taxonomic diversity in oil samples with different degrees of biodegradation and recovered from different reservoirs within a petroleum field of the Campos Basin, Brazil, by using 16S rRNA gene library construction associated with enrichment culture techniques, in order to provide a detailed characterization of the bacterial community and contribute to improve the understanding of biodegradation processes in this environment.
Material and methods
Study area, geological and geochemical background
The Campos Basin covers an area of about 100,000 km2, mostly offshore to the 3,400 m isobath, and is the most prolific oil basin in Brazil, holding approximately 85% of total Brazilian oil reserves and 40% of total gas reserves (Jahnert et al. 1998). The origin of the Campos Basin is related to the Early Cretaceous break up of the Gondwana supercontinent and its tectono-sedimentary evolution can be divided in three main phases: rift where the saline lacustrine source rocks were deposited (Mohiak et al. 1989), transitional that holds the most complex lithology of the sedimentary pile, including siliciclastics, marls, carbonates and evaporites, and drift that began with the sedimentation of shallow carbonates followed by deep water sediments and other shallowing upward sequence (Asmus and Ponte 1973; Ponte and Asmus 1978; Guardado et al. 1989; Rangel et al. 1994).
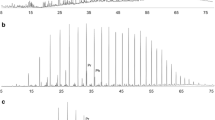
Bulk, isotopic and molecular analyses (Medium Pressure Liquid Chromatography (MPLC); Gas Chromatography (GC); biomarkers; δ13C) of the studied oils have shown diagnostic characteristics of a common source e.g., gammacerane/H30 <0.5; H35/H34 <1.0; very similar ratios among C24 tetracyclic and tricyclic terpanes; similar levels of thermal maturity given by C29 steranes isomers; and different levels of biodegradation (Table 1). The oil samples used in this study were chosen from 73 producing wells within the field, and are representative examples of a non-biodegraded oil (Fig. 1A) produced from the coquina sequence of the Lagoa Feia Formation (≈3,070–3,240 m, reservoir temperature ≈85°C) and of a biodegraded oil (biodegradation level of 5–6 according to Peters and Moldowan 1993; Fig. 1B) produced from sandstones of the Carapebus Formation (≈2,066 m, reservoir temperature ≈51°C).

(A) (1) Gas chromatogram trace of the non-biodegraded oil sample showing low molecular weight n- and isoparaffins, and the relatively low pristane (Pr)/nC17 and phytane (Ph)/nC18. (2) Mass chromatogram (m/z 191) with no significant detection of 25-norhopane. (B) (1) Gas chromatogram (whole oil) of the biodegraded oil sample. Notice the absence of n- and isoparaffins and the significant “hump” representing the unresolved complex mixture (UCM). (2) Mass chromatogram (m/z 191) with a high relative proportion of C29 25-Nor-17α(H)hopane indicated by the “star”. The absence of n- and isoparaffins and the presence of 25-norhopanes characterize the levels of biodegradation 5–6 of Peters and Moldowan (1993)
Sampling
Oil samples were obtained in May of 2002, from a production petroleum platform in the Campos Basin (Brazil). Samples were collected in triplicate using 2 l sterilized Schott bottles, which were completely filled with the samples in order to prevent oxygen influx. Samples were kept on ice during transportation to the laboratory, and stored at 4°C until DNA extraction.
Aerobic microbial enrichments
The oil samples were homogenized using an equal volume (20 ml) of Tween 80 in sterile Falcon tubes and incubated at 40°C for 10 min. Aliquots of 2.5 ml of the oil: tween mixture were inoculated in Falcon tubes containing each 12.5 ml of one of the selected culture media: Nutrient broth (NB Difco), Tripticase soy broth (TSB Difco), Glucose–yeast extract–malt extract (GYM Difco), Marine broth (MB Difco), Acinetobacter broth (Difco) and mineral salt medium Bushnell Hass broth (BHM Difco). The inoculated tubes were incubated at 28°C on a rotary shaker at 150 rpm for 15 days.
Isolation and identification of microbial cultures
Aliquots (100 μl) of each microbial enrichment from crude oils were taken and plated over the surface of the same medium used for enrichment, cited above, supplemented with 2% agar. The plates were inoculated in duplicate and incubated at 28°C and 50°C, under aerobic conditions, up to 10 days. Colony growth was monitored every 2 days. Isolates obtained were further streaked onto the surface of fresh plates (same medium used for enrichment supplemented with 2% agar) and checked for purity prior to subsequent molecular analyses.
DNA extraction
Genomic DNA from the pure cultures was obtained according to the protocol described by Young and Blakesley (1991). DNA extraction from oil samples was carried out by using the QIAmp DNA Stool Mini Kit (Qiagen, Valencia, CA), as previously described by Tanaka et al. (2002) with minor modifications, which included the use of an aliquot of oil correspondent to 2 g of initial sample amount and 30 μl of elution buffer (QIAmp DNA Stool Mini Kit) for final elution of total DNA. Before community DNA extraction, oil samples were heated at 80°C for 30 min in order to reduce the viscosity and allow easier sample manipulation.
PCR amplification
For 16S rRNA gene library construction, amplification was performed from total community DNA isolated from oil samples by using the bacterial primer set 27f and 1100r (Lane 1991). Twenty five microliter reaction mixtures contained 5 μl of total DNA, 2 U of Taq polymerase (GE Healthcare), 0.2 mM of dNTP mix, and 0.4 μM of each primer, in 1× Taq buffer. The PCR amplifications were done using 10 cycles of 1 min at 94°C, 30 s at 60°C, decreasing 0.5°C each cycle, and 3 min at 72°C, followed by another 10 cycles of 1 min at 94°C, 30 s at 56°C and 3 min at 72°C, in an Eppendorf thermal cycler (Eppendorf). A second PCR reaction was performed using 5 μl of the first PCR products as template and the same primers and conditions specified for the first PCR reaction. To overcome known biases introduced by PCR amplification, some procedures reported in previous studies were adopted. The number of PCR cycles was limited to 20 to avoid a reduction in the microbial diversity represented in the libraries (von Wintzingerode et al. 1997; Schink 1997) and to minimize alterations in community composition caused by “bias 1:1” (Watanabe et al. 2002), which can result in higher equitability in the final amplified community. In addition, the products of five PCR amplification replicates were pooled prior to cloning in order to minimize PCR drift (Friedrich et al. 1997; Munson et al. 1997).
Cloning, ARDRA analysis and sequencing
A 16S rRNA gene library was constructed for each of two replicate oil samples: NBD library originated from the non-biodegraded oil, and HBD library originated from the high-biodegraded oil. Amplicons were pooled from five reactions (∼500 ng), purified using GFX TM PCR-DNA and gel band purification kit (GE Healthcare) and cloned using the pGEM-T cloning vector kit, according to the manufacturer’s instructions (Promega, Madison, Wisc.). E. coli DH5α cells were transformed by electroporation, using the equipment Cellject Uno (Hybaid). White colonies were selected and screened for the presence of insert by means of PCR amplification using the vector-based primers M13 forward and reverse. All the insert-containing clones were submitted to Amplified Ribosomal DNA Restriction Analysis (ARDRA) by digestion with the enzymes HaeIII and RsaI (GE Healthcare), at 37°C for 4 h. The fragments were separated by electrophoresis in 2.5% agarose gels and the different ribotypes were defined by a combination of the banding patterns obtained with the two enzymes. One clone representative of each distinct ribotype was selected for DNA sequencing and phylogenetic affiliation. Plasmid DNA was isolated from clones using the protocol described by Sambrook et al. (1989) and purified with the GFX PCR DNA and gel band purification kit (GE Healthcare) prior to sequencing. The 16S rRNA gene sequences were determined from plasmid DNA by using the M13 forward and reverse primers and the DYEnamic ET Dye Terminator Cycle Sequencing Kit for the automated MegaBace 500 system (GE Healthcare), according to the manufacturer’s recommendations.
Identification of bacterial isolates
PCR amplification of 16S rRNA gene fragments from pure cultures and subsequent direct sequencing in a MegaBace 500 system (GE Healthcare), aiming at the phylogenetic affiliation of the isolates, were performed as described by Rainey et al. (1996).
Phylogenetic analysis
Partial 16S rRNA gene sequences obtained from clones or pure cultures using forward and reverse primers were assembled in a contiguous sequence using the phred/Phrap/CONSED program (http://www.phrap.org/phredphrapconsed.html). Identification was achieved by comparing the contiguous 16S rRNA gene sequences (∼1000 bp in length) generated for clones and isolates with 16S rRNA sequence data from reference and type strains, as well as environmental clones, available at the public databases Genbank (http://www.ncbi.nem.nih.gov) and the RDP (Ribosomal Database Project, Wiscosin, USA, http://www.cme.msu.edu/RDP/html/index.html) by using the BLASTN and the RDP sequence match routines. The sequences were aligned using the CLUSTAL X program (Thompson et al. 1994) and analyzed using PAUP (version 4.0 beta 10) (Swofford 2000). Evolutionary distances were derived from sequence-pair dissimilarities calculated as implemented in PAUP, using Kimura 2P substitution model (Kimura 1980). The phylogenetic reconstruction was done using the neighbor-joining (NJ) algorithm, with bootstrap values calculated from 1,000 replicate runs, using the routines included in the PAUP software.
Statistical analyses based on ARDRA data
Randomization analyses based on ARDRA data were performed using the independent sampling algorithm, implemented in the EcoSim program (Gotelli and Entsminger 2003). This module of EcoSim provides a computer-sampling algorithm, in which a specified number of individuals (i.e., sub-samples of the original one) are randomly drawn, without replacement, from the total community sample, creating pseudo-communities. Various sub-sample sizes were specified and for each sub-sample size, 1000 pseudo-communities were drawn from the original one. Data from these were used to calculate diversity indices such as Shannon–Wiener index (diversity), Dominance Index and PIE Hulbert’s index (evenness), as well as richness. The resultant values were used to calculate the mean and the 95% confidence intervals for each index at each sub-sample size specified. This sampling algorithm ensures that the estimators of diversity parameters for the different sub-sample efforts are independent of one another.
Nucleotide sequence accession numbers
The nucleotide sequences determined in this study for the environmental clones were deposited at the Genbank database under the accession numbers: AY862487–AY862494 (Fig. 3). For the bacterial isolates, the accession numbers are: DQ413027–DQ413031.
Results
NBD and HBD 16S rRNA gene libraries
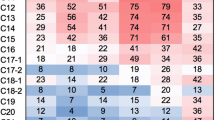
PCR amplification of 16S rRNA gene fragments from community DNA obtained from oil samples was successful only when applying a second round of amplification, using the PCR products from a first PCR as template. A total of 137 and 129 clones were obtained for the NBD and HBD oil libraries, respectively. ARDRA analysis showed 46 different restriction patterns (ribotypes) for the NBD library and 35 for the HBD library (Table 2). One representative of each ribotype, considering both libraries, was sequenced. BLASTN analysis of clone sequences revealed that they belonged to nine different genera (Table 2, Fig. 2). One clone representative of each taxonomic group, considering both NBD and HBD libraries, was used for the phylogenetic tree reconstruction (bo22C02, bo23A06, bo13A01, bo13D04, bo22B07, bo22A03b, bo13A02b and bo13C09) (Fig. 3), except for the clone representing the genus Leuconostoc (bo12F03), which was not included in the phylogenetic analysis due to its low quality 16S rRNA gene sequence.

Richness and frequency of ARDRA patterns found in the NBD and HBD 16S rDNA libraries. Different segments in the bars indicate distinct ARDRA patterns (ribotypes)

Neighbor-joining tree showing the phylogenetic relationships between the environmental clones derived from NBD and HBD libraries and related organisms
Sequence analysis revealed that 16S rRNA gene clones were related to the genera Acinetobacter, Arcobacter, Bacillus, Halanaerobium, Leuconostoc, Marinobacter, Propionibacterium, Streptococcus and Streptomyces, encompassing four main groups within the Domain Bacteria: Actinobacteria (high-G + C gram-positive bacteria), Firmicutes (low-G + C gram-positive bacteria), gamma-Proteobacteria and epsilon-Proteobacteria (Fig. 3). Arcobacter, Halanaerobium, Marinobacter and Propionibacterium were found in both NBD and HBD libraries, whereas Leuconostoc and Streptomyces were found only in the NBD library, and Acinetobacter, Bacillus and Streptococcus only in the HBD library (Fig. 2).
Ribotypes related to the genus Halanaerobium were the most abundant in the NBD library (61.31%), followed by the ribotypes related to Arcobacter (22.62%) (Table 2, Fig. 2). On the other hand, the genera Propionibacterium and Acinetobacter were the most abundant amongst sequences recovered from the biodegraded oil (HBD), representing 50.38% and 31.58% of the clones in the library, respectively (Table 2, Fig. 2).
Amongst the four bacterial groups found in both NBD and HBD libraries, the low-G + C gram-positive bacteria was the predominant one, and accounted for 38.71% of the total clones in the libraries. Clones representing this group showed high sequence similarity with Bacillus sporothermodurans type strain (bo22C02, 100% bootstrap value), Streptococcus sanguinis (bo23A06, 99% bootstrap value) and Halanaerobium congolense type strain (bo13A01, 100% bootstrap value) (Fig. 3). The class Actinobacteria accounted for 31.56% of the total number of clones, with clones bo13D04 and bo22B07 representative of the two genera found for this class. Clone bo13D04 grouped with Streptomyces albidoflavus and Streptomyces somalienses, with 100% bootstrap value, whereas clone bo22B07 formed a tight cluster (supported by 82% bootstrap value) with Propionibacterium acnes type strain and two other Propionibacterineae, which were uncultured bacteria from hypersaline lake and hydrocarbon seep, respectively. The two other groups, gamma-Proteobacteria and epsilon-Proteobacteria, accounted for 17.28% and 12.03% of all clones, respectively. Clones bo22A03b and bo13A02b, representative of gamma-Proteobacteria, were related to Acinetobacter lwoffii (72% bootstrap value) and to Marinobacter lipolyticus type strain (100% bootstrap value). Clone bo13C09, representative of epsilon-Proteobacteria, showed high sequence similarity with a solar lake Arcobacter sp. (100% bootstrap value) (Fig. 3).
Statistical comparisons of libraries based on ARDRA data
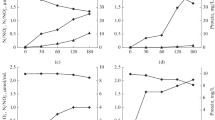
In order to investigate the community structures represented in the two libraries, diversity indices were used to calculate richness, diversity, dominance and evenness from the ARDRA data. Since these indices are dependent on the total number of clones analyzed and the two libraries were unequally sampled (137 clones in NBD and 129 in HBD), randomization analyses were used to create pseudo-communities at specified sub-sample sizes and thus allow the comparison between the two libraries. Relatively high ribotype richness, as well as high diversity index (Shannon–Wiener), values were observed for both library communities (Fig. 4A, B). The communities from NBD and HBD did not present statistically different diversity index values based upon HaeIII/RsaI ARDRA data at all the sub-sample sizes analyzed (Fig. 4B). The richness analysis showed a slightly different pattern, at least at the larger sub-sample sizes. This data indicates that, when analyzing sample sizes with at least 100 individuals, the community represented in the NBD library may show significantly higher number of different ribotypes than the HBD library or, in other words, higher richness (Fig. 4A). The PIE Hulbert’s evenness index (Fig. 4C) calculates the probability of an inter-specific encounter, i.e., the probability that two randomly sampled individuals from a community represent two different ribotypes. High values for PIE index were reached very rapidly in the analyses indicating that both communities contained a large proportion of singletons (ribotype of single occurrence) (Fig. 4C). Evenness index values did not show statistically significant differences between NBD and HBD libraries, at all sub-sample sizes analyzed. The dominance index, the fraction of the community that is represented by the most common ribotype, reached relatively low values for both communities (Fig. 4D), thus revealing that these did not present strong dominance of a particular ribotype. However, when analyzing sample sizes with at least 80–90 individuals, the community represented in the NBD library may show significantly higher dominance values (Fig. 4D).

Statistical comparisons of libraries based on ARDRA data. NBD library (■) and HBD library (□)
Enrichment and isolation of aerobic microorganisms from crude oil
Five different bacterial genera were recovered from the biodegraded oil sample using cultivation methods. All the isolates obtained were recovered from the enrichments incubated at 28°C, which were subsequently identified and deposited at the Brazilian Collection of Environmental and Industrial Microorganisms (CBMAI). Gram positive rods were observed in the NB culture medium, which were identified as Bacillus subtillis (CBMAI 707) based on 16S rDNA sequence analysis. Gram negative rods were observed on TSB medium, identified as Methylobacterium sp. (CBMAI 706). In the Acinetobacter broth medium, Gram negative coccal rods were recovered and identified as Achromobacter xylosoxidans (CBMAI 709), and when using MB medium, Gram positive long rods identified as Brevibacillus sp. (CBMAI 708) were obtained. In the salts medium Gram positive coco bacilli were observed and characterized as Dietzia sp. (CBMAI 705). No growth was observed in the media PDB and GYM.
Discussion
The nature and diversity of bacteria in oil field ecosystems is still poorly understood. However, recent incorporation of molecular methods has allowed a broader characterization of microbial assemblages in this type of environment. The majority of studies in petroleum microbiology have been conducted using samples of formation water or water–oil mixtures. Only one study reported in the literature used crude oil from petroleum reservoir to analyze microbial communities (Tanaka et al. 2002).
In our study, data derived from cloning, sequencing and phylogenetic analyses allowed, for the first time, the comparison of bacterial communities from oil samples with different levels of biodegradation, originated from Brazilian petroleum reservoirs. However, in order to properly interpret the data obtained, one must keep in mind the limitations inherent to the methodological approaches used. These include the potential impact of sample heating at 80°C, prior to DNA extraction, on the original bacterial composition of the biodegraded oil, considering that the reservoir temperature, in this case, is about 51°C.
All clones obtained from NBD and HBD 16S rRNA gene libraries belonged to four major bacterial classes: Firmicutes, Actinobacteria, gamma-Proteobacteria and epsilon-Proteobacteria, revealing a great microbial diversity in these oil samples.
Sequence analysis of clones from the NBD and HBD libraries revealed phylogenetic relationships with 16S rRNA gene sequences from many microorganisms previously isolated or identified from petroleum, marine and saline lake habitats, such as Acinetobacter (Saadoun 2002), Halanaerobium (Ravot et al. 1997), Marinobacter (Huu et al. 1999) and Arcobacter (Watanabe et al. 2000). Other groups that showed similarity with some of the sequenced clones are not mainly found in these habitats, but according to the literature, they may have some representatives found in oil environments or related to hydrocarbon degradation. This is the case for the genera Bacillus (Stapleton et al. 2000; Nazina et al. 2002; Zhuang et al. 2003), Propionibacterium (Yoshida et al. 2005; Vorob’eva et al. 1979) and Streptomyces (Radwan et al. 1995, 1998; Bachoon et al. 2001). However, the genera Leuconostoc and Streptococcus are usually associated with dairy foods (Damelin et al. 1995; Stiles and Holzapfel 1997) and clinical pathology (Tyrrell et al. 2005), respectively, and this is the first report of the presence of these bacteria associated to oil field or marine environment.
Some of these microbial groups were shown to be exclusive of the biodegraded oil sample, such as the genera Acinetobacter, Bacillus and Streptococcus. Amongst these, Acinetobacter was the most abundant and it has been recognized in the literature as an aerobic degrader of hydrocarbon and lipids (Bach et al. 2003; Pleshakova et al. 2001; Sugimori et al. 2002). In the phylogenetic tree, the clone bo22A03b was shown to be related to A. lwofii, Acinetobacter baumannii, Acinetobacter calcoaceticus and to one unidentified Acinetobacter strain from deep subsurface environment (Vepritskiy et al. 2002). According to Barkovskii and Shub (1986) A. calcoaceticus is able to degrade a dense spectrum of aromatic compounds.
Members from Bacillus have been associated with petroleum and hydrocarbon degradation in many others studies (Stapleton et al. 2000; Nazina et al. 2002; Saadoun 2002; Bach et al. 2003; Lu et al. 2003; Zhuang et al. 2003). Clone bo22C02 showed a close relationship with B. sporothermodurans type strain, which is able to produce highly heat-resistant endospores (Pettersson et al. 1996), and with two others species, Bacillus sonorensis and Bacillus licheniformis, the latter being both thermophilic and halotolerant (Batrakov et al. 2003).
Genera Arcobacter, Halanaerobium, Marinobacter and Propionibacterium were found in both 16S rRNA gene libraries (NBD and HBD) constructed from oil samples. Members from the genus Arcobacter are nitrate-reducing and sulfide-oxidizing bacteria, and some of them have been found in marine and lake habitats, such as the Hawaiian Archipelago, Niger Sea sediments (Thamdrup et al. 2000), North Sea (Eilers et al. 2000) and Solar Lake (Teske et al. 1996). Arcobacter spp. has also been isolated from oil field (Gevertz et al. 2000).
Mesophilic fermentative bacteria, such as the genus Halanaerobium, together with thermophilic and hyperthermophilic fermentative bacteria, constitute an important microbial community of the oil field environment (Magot et al. 2000). Three species of haloanaerobes isolated from oil field brines, Halanaerobium acetoethilicum, Halanaerobium congolense (Ravot et al. 1997) and Halanaerobium salsugo (Bhupathiraju et al. 1994) are heterotrophic moderate halophyles that can use carbohydrates to produce metabolites (acids, solvents and gases), and may be of interest for potential use in microbial enhancement of oil recovery (MEOR). In addition, Ravot et al. (1997) reported that H. congolense was able to reduce thiosulfate to H2S, a compound known to decrease oil quality, corrode steel material and threaten worker’s health due to its high toxicity (Magot et al. 2000).
Representatives from genus Marinobacter are, in general, facultative anaerobes, thermophilic and halophilic organisms. M. lipolyticus, which showed the closest relationship with clone bo13A02b, is a halophylic organism that possesses lipolytic activity. Two other species grouped with clone bo13A02b, Marinobacter hydrocarbonoclasticus, a marine bacterium highly halotolerant and able to degrade hydrocarbon (Gauthier et al. 1992; Doumenq et al. 2001; Barbeau et al. 2002), and Marinobacter aquaeolei, a bacterium isolated from a Vietnamese oil-producing well (Huu et al. 1999).
Clone bo22B07 was related to the P. acnes type strain and to two uncultured Propionibacterineae bacteria, found in hypersaline lakes and hydrocarbon seeps. The presence of Propionibacterium in high abundance in the biodegraded oil (Fig. 2, Table 2) is corroborated by the findings of Yoshida and co-workers (2005), who detected P. acnes at high frequency in samples from stored crude oil, suggesting that these bacteria are typical from crude oil and not contaminants of the oil stockpile. Yet, this genus may be related to lipolytic activity (Jarvis et al. 1998) or hydrocarbon oxidation (Vorob’eva et al. 1979).
Although Streptomyces spp. are well recognized as hydrocarbon degraders (Radwan et al. 1995, 1998; Bachoon et al. 2001), their abundance was shown to be higher in the NBD library (Fig. 2). This might be explained by the fact that these organisms are not the main responsible for hydrocarbon degradation in these oil fields. In the phylogenetic tree, clone bo13D04 formed a tight cluster with S. albidoflavus and S. somaliensis species (Fig. 3). This group showed close relationship with a thermotolerant Streptomyces strain and with an isolate from marine sediments. The other specific NBD library genus was Leuconostoc, represented by only one clone related to Leuconostoc citreum. This genus has already been related with MEOR (Behlülgil and Mehmetogelu 2002).
Randomization analysis of ARDRA data revealed valuable information about the genetic structure of the communities recovered in the NBD and HBD libraries. Diversity and evenness analyses showed similar community structures in both libraries, with no significant differences for all sub-samples analyzed. The high PIE Hulbert Index values observed (Fig. 4C), reaching almost the maximum value (indicative of high proportion of singletons), and the steep shape of the richness curve (Fig. 4A), strongly indicates that much more diversity may be encountered if larger sample efforts are made. These data also show that there is a high genetic diversity in the bacterial communities inhabiting both environments, with no correlation to the oil biodegradation level. Despite the high evenness of both communities, these showed some dominance of the most common ribotype, expressed by the stabilization of the dominance index at values between 0.16 and 0.27 (Fig. 4D). The difference observed between the indices from both libraries could be explained by the high abundance of one of the ribotypes representing the Halanaerobium genus in the NBD library (Fig. 2).
It is important to note that due to the nature of the randomization analyses used, comparisons can be made only in relation to the sample effort of the smallest sample, which is 129 clones (from HBD library), and should not be extrapolated to larger samples. Moreover, these comparisons relate just to the samples analyzed and cannot be extrapolated to the original community from which the libraries were constructed.
Parallel experiments carried out by the authors have employed cultivation-based techniques in order to evaluate the diversity of cultivable bacteria in the same samples. The strategy involved the use of culture media for enrichment of heterotrophic aerobic bacteria, using oil samples as inoculum and sole carbon source. 16S rRNA gene sequencing-based identification of the isolated microorganisms showed the presence of bacteria related to A. xylosoxidans, B. subtilis, Brevibacillus sp., Dietzia sp. and Methylobacterium sp. These organisms have all been previously reported in the literature as hydrocarbon degraders and/or associated to oil field environments (Al-Sharidah et al. 2000; Bieszkiewicz et al. 1998; Chaillan et al. 2004; Orphan et al. 2000; Pineda-Flores et al. 2004; Nazina et al. 2002), suggesting that they might have a widespread distribution in oil reservoirs.
Comparison between the data obtained using cultivation-independent and culture-based enrichment methods suggests that different selection of community members may occur. Only the genus Bacillus was recovered using both strategies. This data may indicate that microorganisms recovered by enrichment cultures and not detected by molecular methods are not major components of the in situ reservoir assemblage. Alternatively, they may have been missed due to PCR biases when using community DNA, such as preferential amplification, or differential lysis efficiency may have occurred when purifying DNA from the samples prior to 16S rRNA gene library assembly.
Theses discrepancies between microbial groups recovered using the two strategies were also observed by other authors in different environments, such as soil and sediment (Chandler et al. 1997; Stephen et al. 1996), suggesting that a more comprehensive assessment of microbial diversity in oil, and probably other environments, can be obtained by using a combination of culture- and molecular-based techniques than by using either method alone.
In conclusion, analysis of bacterial communities in oil samples from Brazilian reservoirs by using a combined approach, 16S rRNA gene libraries and enrichment cultures, was successful and allowed the identification of distinct microbial groups known to be associated with petroleum and/or marine environments. This study presents an emerging view of the bacterial community diversity of Brazilian oil samples, based on both molecular and cultivation techniques, allowing researchers insights into the microorganisms that might be involved in the biogeochemical transformations that take place in these environments.
References
Al-Sharidah A, Richardt A, Golecki JR, Dierstein R, Tadros MH (2000) Isolation and characterization of two hydrocarbon-degrading Bacillus subtilis strains from oil contaminated soil of Kuwait. Microbiol Res 155:157–164
Asmus HE, Ponte FC (1973) The Brazilian marginal basins. In: Nairn AE, Sthli FG (eds) The ocean basins and margins, vol. 1, South Atlantic, Plenun Press, New York, pp 87–133
Bach H, Berdichevsky Y, Gutnick D (2003) An exocellular protein from the oil-degrading microbe Acinetobacter venetianus RAG-1 enhances the emulsifying activity of the polymeric bioemulsifier emulsan. Appl Environ Microbiol 69:2608–2615
Bachoon DS, Araujo R, Molina M, Hodson RE (2001) Microbial community dynamics and evaluation of bioremediation strategies in oil-impacted salt marsh sediment microcosms. J Ind Microbiol Biotechnol 27:72–79
Barbeau K, Zhang G, Live DH, Butler A (2002) Petrobactin, a photoreactive siderophore produced by the oil-degrading marine bacterium Marinobacter hydrocarbonoclasticus. J Am Chem Soc 124:378–379
Barkovskii AL, Shub GM (1986) Acinetobacter calcoaceticus strain with a wide spectrum of utilizing aromatic compounds and carrying a plasmid for resorcin degradation. Mikrobiologiia 55(2):237–240
Batrakov SG, Rodionova TA, Esipov SE, Polyakov NB, Sheichenko VI, Shekhovtsova NV, Lukin SM, Panikov NS, Nikolaev YA (2003) A novel lipopeptide, an inhibitor of bacterial adhesion, from the thermophilic and halotolerant subsurface Bacillus licheniformis strain 603. Biochim Biophys Acta 1634(3):107–150
Behlülgil K, Mehmetogelu MT (2002) Bacteria for improvement of oil recovery: a laboratory study. Energy Sources 24:413–421
Bieszkiewicz E, Horoch M, Boszczyk-Maleszak H, Mycielski R (1998) An attempt to use selected strains of bacteria adapted to high concentrations of petroleum oil to increase the effective removal of petroleum products in excess activated sludge in laboratory conditions. Acta Microbiol Pol 47:305–312
Bhupathiraju VK, Oren A, Sharma PK, Tanner RS, Woese CR, McInerney MJ (1994) Haloanaerobium salsugo sp. nov., a moderately halophilic, anaerobic bacterium from a subterranean brine. Int J Syst Bacteriol 44(3):565–572
Bonch-Osmolovskaya EA, Miroshnichenko ML, Lebedinsky AV, Chernyh NA, Nazina TN, Ivoilov VS, Belyaev SS, Boulygina ES, Lysov YP, Perov AN, Mirzabekov AD, Hippe H, Stackebrandt E, L’Haridon S, Jeanthon C (2003) Radioisotopic, culture-based, and oligonucleotide microchip analyses of thermophilic microbial communities in a continental high-temperature petroleum reservoir. Appl Environ Microbiol 69:6143–6151
Chaillan F, Le Fleche A, Bury E, Phantavong YH, Grimont P, Saliot A, Oudot J (2004) Identification and biodegradation potential of tropical aerobic hydrocarbon-degrading microorganisms. Res Microbiol 155:587–595
Chandler DP, Shu-Mei L, Spadoni CM, Drake GR, Balkwill DL, Fredrickson JK, Brockman FJ (1997) A molecular comparison of culturable aerobic heterotrophic bacteria and 16S rDNA clones derived from a deep subsurface sediment. FEMS Microbiol Ecol 23:131–144
Connan J (1984) In: Brooks J, Welte DH (eds) Advances in petroleum geochemistry, vol 1. Academic, London, pp 299–335
Damelin LH, Dykes A, von Holy A (1995) Biodiversity of lactic acid bacteria from food-related ecosystems. Microbios 83:13–22
DeLong EF (1992) Archaea in coastal marine environments. Proceedings of the Natural Academy of Sciences USA 89:5685–5689
Doumenq P, Aries E, Asia L, Acquaviva M, Artaud J, Gilewicz M, Mille G, Bertrand JC (2001) Influence of n-alkanes and petroleum on fatty acid composition of a hydrocarbonoclastic bacterium: Marinobacter hydrocarbonoclasticus strain 617. Chemosphere 44:519–528
Eilers H, Pernthaler J, Glockner FO, Amann R (2000) Culturability and in situ abundance of pelagic bacteria from the North Sea. Appl Environ Microbiol 66(7):3044–3051
Gauthier MJ, Lafay B, Christen R, Fernandez L, Acquaviva M, Bonin P, Bertrand JC (1992) Marinobacter hydrocarbonoclasticus gen. nov., sp. nov., a new, extremely halotolerant, hydrocarbon-degrading marine bacterium. Int J Syst Bacteriol 42(4):568–576
Gevertz D, Telang AJ, Voordouw G, Jenneman GE (2000) Isolation and characterization of strains CVO and FWKO B, two novel nitrate-reducing, sulfide-oxidizing bacteria isolated from oil field brine. Appl Environ Microbiol 66(6):2491–2501
Gotelli NJ, Entsminger GL (2003) EcoSim: null models software for ecology. Acquired Intelligence Inc. & Kesey-Bear, Burlington, VT 05465
Guardado LR, Gamboa LAP, Lucchesi CF (1989) Petroleum geology of the Campos Basin, Brazil, a model for a producing Atlantic type basin. In: Edwards JD, Santogrossi PA (eds) Divergent/passive margin basins, AAPG Memoir 48, 3–79
Head IM, Jones DM, Larter SR (2003) Biological activity in the deep subsurface and the origin of heavy oil. Nature 426:344–352
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Huu NB, Denner EB, Ha DT, Wanner G, Stan-Lotter H (1999) Marinobacter aquaeolei sp. nov., a halophilic bacterium isolated from a Vietnamese oil-producing well. Int J Syst Bacteriol 49(2):367–375
Jahnert R, França A, Trindade LAF, Quintaes C, Santos P, Pessoa J, Bedregal RP (1998) The petroleum system of Campos Basin. BGP; AAPG International Conference & Exhibition, November 8–11, 1998, Rio de Janeiro, Brasil. Extended abstracts volume, 600–601
Jarvis GN, Strompl C, Moore ER, Thiele JH (1998) Isolation and characterisation of obligately anaerobic, lipolytic bacteria from the rumen of red deer. Syst Appl Microbiol 21:135–143
Juck D, Charles T, Whyte LG, Greer CW (2000) Polyphasic microbial community analysis of petroleum hydrocarbon-contaminated oils from two northern Canadian communities. FEMS Microbiol Ecol 33:241–249
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Lane DJ (1991) 16S/23S rRNA Sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Chichester, England, pp 115–175
Larter S, Wilhelms A, Head IM, Koopmans M, Aplin R, Di Primio R, Zwach C, Erdmann M, Telnaes N (2003) The controls on the composition of biodegraded oils in the deep subsurface – part 1: biodegradation rates in petroleum reservoirs. Org Geochem 34:601–613
Larter S, Huang H, Adams J, Bennett B, Jokanola O, Oldemburg T, Jones M, Head I, Riediger C, Fowler M (2006) The controls on the composition of biodegraded oils in the deep subsurface – part II: geological controls on subsurface biodegradation fluxes and constraints on reservoir fluid property prediction. AAPG Bull 90:921–938
Lu XX, Zhang X, Li GH, Zhang WH (2003) Production of biosurfactant and its role in the biodegradation of oil hydrocarbons. J Environ Sci Health A Tox Hazard Subst Environ Eng 38:483–492
Magot M, Ollivier B, Patel BKC (2000) Microbiology of petroleum reservoirs. Ant Van Leeuwen 77:103–116
Mohiak WU, Mello MR, Karner GD, Dewey JF, Maxwell JR (1989) Structural and stratigraphic evolution of the Campos Basin, offshore Brazil. Extentional Tectonics and Stratigraphy of North Atlantic Margins: Analogs, AAPG Memoir 46:577–598
Munson MA, Nedwell DB, Embley TM (1997) Phylogenetic diversity of Archaea in sediment samples from a coastal salt marsh. Appl Environ Microbiol 63:4729–4733
Nazina TN, Grigor’ian AA, Sue KF, Sokolova DSh, Novikova EV, Turova TP, Poltaraus AB, Beliaev SS, Ivanov MV (2002) Phylogenetic diversity of aerobic saprotrophic bacteria isolated from the Daqing oil field. Mikrobiologiia 71:103–110
Orphan VJ, Taylor LT, Hafenbradl D, Delong EF (2000) Culture-dependent and culture-independent characterization of microbial assemblage associated with high temperature petroleum reservoirs. Appl Environ Microbiol 66:700–711
Pace NR (1996) New perspective on the natural microbial world: molecular microbial ecology. ASM News 62:463–470
Peters KE, Moldowan JM (1993) The Biomarkers Guide. Prentice-Hall, New York, 363 pp
Pettersson B, Lembke F, Hammer P, Stackebrandt E, Priest FG (1996) Bacillus sporothermodurans, a new species producing highly heat-resistant endospores. Int J Syst Bacteriol 46(3):759–764
Pineda-Flores G, Boll-Arguello G, Lira-Galeana C, Mesta-Howard AM (2004) A microbial consortium isolated from a crude oil sample that uses asphaltenes as a carbon and energy source. Biodegradation 15:145–151
Pleshakova EV, Muratova AIu, Turkovskaia OV (2001) Degradation of mineral oil by Acinetobacter calcoaceticus strain. Prikl Biokhim Mikrobiol 37:398–404
Ponte FC, Asmus HE (1978) Geological framework of the Brazilian continental margin. Geol Rundsch 67:201–235
Radwan SS, Sorkhoh NA, Fardoun F, Al-Hasan RH (1995) Soil management enhancing hydrocarbon biodegradation in the polluted Kuwaiti desert. Appl Microbiol Biotechnol 44(1–2):265–270
Radwan SS, Barabas G, Sorkhoh NA, Damjanovich S, Szabo I, Szollosi J, Matko J, Penyige A, Hirano T, Szabo IM (1998) Hydrocarbon uptake by Streptomyces. FEMS Microbiol Lett 169(1):87–94
Rainey FA, Ward-Reiney N, Kroppenstedt RM, Stackebrandt E (1996) The genus Nocardiopsis represents a phylogenetically coherent taxon and a distinct actinomycete lineage: Proposal of Nocardiopsaceae fam. nov. Int J Syst Bacteriol 46:1088–1092
Rangel HD, Martins FAL, Esteves FR, Feijó FJ (1994) Bacia de Campos. Boletim de Geociências da Petrobras 8(1):203–217
Ravot G, Magot M, Ollivier B, Patel BK, Ageron E, Grimont PA, Thomas P, Garcia JL (1997) Haloanaerobium congolense sp. nov., an anaerobic, moderately halophilic, thiosulfate- and sulfur-reducing bacterium from an African oil field. FEMS Microbiol Lett 147(1):81–88
Saadoun I (2002) Isolation and characterization of bacteria from crude petroleum oil contaminated soil and their potential to degrade diesel fuel. J Basic Microbiol 42:420–428
Sambrook J, Fritsch EF, Maniatis T (1989) Small-scale preparations of plasmid DNA. In: Molecular cloning: a laboratory manual, 2nd edn., vol 1. Cold Spring Harbor Laboratory Press, Plainview, NY, pp 25–30
Schink B (1997) Energetics of syntrophic cooperation in methanogenic degradation. Microbiol Mol Biol Rev 61:262–280
Stapleton RD, Bright NG, Sayler GS (2000) Catabolic and genetic diversity of degradative bacteria from fuel-hydrocarbon contaminated aquifers. Microb Ecol 39(3):211–221
Stephen JR, McCaig AE, Smith Z, Prosser JI, Embley TM (1996) Molecular diversity of soil and marine 16S rRNA gene sequences related to beta-subgroup ammonia-oxidizing bacteria. Appl Environ Microbiol 62:4147–4154
Stiles ME, Holzapfel WH (1997) Lactic acid bacteria for foods and their current taxonomy. Review article Int J Food Microbiol 36:1–29
Sugimori D, Nakamura M, Mihara Y (2002) Microbial degradation of lipid by Acinetobacter sp. strain SOD-1. Biosci Biotechnol Biochem 66(7):1579–1582
Swofford DL (2000) PAUP*. Phylogenetic Analysis Using Parsimony (*and other methods). Sinauer Associates, Sunderland, MA
Takami H, Inoue A, Fuji F, Horikoshi K (1997) Microbial flora in the deepest sea mud of the Mariana Trench. FEMS Microbiol Lett 152:279–285
Tanaka Y, Sogabe M, Okumura K, Kurane R (2002) A highly selective direct method of detecting sulphate-reducing bacteria in crude oil. Lett Appl Microbiol 35:242–246
Teske A, Sigalevich P, Cohen Y, Muyzer G (1996) Molecular identification of bacteria from a coculture by denaturing gradient gel electrophoresis of 16S ribosomal DNA fragments as a tool for isolation in pure cultures. Appl Environ Microbiol 62(11):4210–4215
Thamdrup B, Rossello-Mora R, Amann R (2000) Microbial manganese and sulfate reduction in Black Sea shelf sediments. Appl Environ Microbiol 66(7):2888–2897
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tyrrell GJ, Lovgren M, Kress B, Grimsrud K (2005) Invasive Group A streptococcal disease in Alberta, Canada, 2000–2002. J Clin Microbiol 43:1678–1683
Van Hamme JD, Singh A, Ward OP (2003) Recent advances in petroleum microbiology. Microbiol. Mol. Biol. Rev. 67:503–549
Vepritskiy AA, Vitol IA, Nierzwicki-Bauer SA (2002) Novel group I intron in the tRNA(Leu)(UAA) gene of a gamma-proteobacterium isolated from a deep subsurface environment. J Bacteriol 184:1481–1487
von Wintzingerode F, Goebel UB, Stackebrandt E (1997) Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21:219–233
Vorob’eva LI, Kraeva NI, Ebringer L, Ol’sinskaia NL (1979) n-Alkane oxidation by propionic acid bacteria. Mikrobiologiia 48:33–38
Voordouw G, Armstrong SM, Reimer MF, Fouts B, Telang AJ, Shen Y, Gevertz D (1996) Characterization of 16S rRNA genes from oil field microbial communities indicates the presence of a variety of sulfate-reducing, fermentative, and sulfide-oxidizing bacteria. Appl. Environ. Microbiol. 62:1623–1629
Watanabe K, Watanabe K, Kodama Y, Syutsubo K, Harayama S (2000) Molecular characterization of bacterial populations in petroleum-contaminated groundwater discharged from underground crude oil storage cavities. Appl Environ Microbiol 66:4803–4809
Watanabe K, Kodama Y, Hamamura N, Kaku N (2002) Diversity, abundance, and activity of archaeal populations in oil-contaminated groundwater accumulated at the bottom of an underground crude oil storage cavity. Appl Environ Microbiol 68:3899–3907
Yoshida N, Yagi K, Sato D, Watanabe N, Kuroishi T, Nishimoto K, Yanagida A, Katsuragi T, Kanagawa T, Kurane R, Tani Y (2005) Bacterial communities in petroleum oil in stockpiles. J Biosci Bioeng 99:143–149
Young A, Blakesley R (1991) Sequencing plasmids form single colonies with the dsDNA cycle sequencing system. Focus 13:13
Zhuang WQ, Tay JH, Maszenan AM, Krumholz LR, Tay ST (2003) Importance of Gram-positive naphthalene-degrading bacteria in oil-contaminated tropical marine sediments. Lett Appl Microbiol 36:251–257
Acknowledgments
L.D. Sette was supported by grants from CNPq Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil. We thank to PETROBRAS for the financial support and authorization to publish this work, to J.R. Cerqueira for his insightful suggestions, and to Dr. Fernanda F. Piza for the critical reading.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sette, L.D., Simioni, K.C.M., Vasconcellos, S.P. et al. Analysis of the composition of bacterial communities in oil reservoirs from a southern offshore Brazilian basin. Antonie van Leeuwenhoek 91, 253–266 (2007). https://doi.org/10.1007/s10482-006-9115-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-006-9115-5