
Abstract
The Notch signalling pathway is one of the main regulators of endothelial biology. In the last 20 years the critical function of Notch has been uncovered in the context of angiogenesis, participating in tip-stalk specification, arterial-venous differentiation, vessel stabilization, and maturation processes. Importantly, pharmacological compounds targeting distinct members of the Notch signalling pathway have been used in the clinics for cancer therapy. However, the underlying mechanisms that support the variety of outcomes triggered by Notch in apparently opposite contexts such as angiogenesis and vascular homeostasis remain unknown. In recent years, advances in -omics technologies together with mosaic analysis and high molecular, cellular and temporal resolution studies have allowed a better understanding of the mechanisms driven by the Notch signalling pathway in different endothelial contexts. In this review we will focus on the main findings that revisit the role of Notch signalling in vascular biology. We will also discuss potential future directions and technologies that will shed light on the puzzling role of Notch during endothelial growth and homeostasis. Addressing these open questions may allow the improvement and development of therapeutic strategies based on modulation of the Notch signalling pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Our body contains an extensive tubular network of blood vessels lined by a monolayer of endothelial cells (ECs) that arose during evolution in order to carry oxygen and nutrients to distant tissues and organs in the body [1]. The first vascular structures are assembled by coalescence of endothelial progenitor cells and in situ differentiation, a process known as vasculogenesis, which gives rise to the first arteries (dorsal aorta), veins and the primitive vascular plexus [2]. This primary vessel network is later expanded through the process of angiogenesis that requires an equilibrium between endothelial cell proliferation, sprouting and migration in order to form new blood vessels and at the same time maintain the function of pre-existing ones. Angiogenesis involves not only the formation of new blood vessels, but also the remodelling and arteriovenous differentiation of pre-existing blood vessels, in order to maintain an effective vascular function that fulfils the metabolic needs of the surrounding tissues. This involves a constant cross-talk between developing or remodelling blood vessels and the surrounding tissues. Over time, a given tissue vascular bed will adopt different architectures to match the evolving demands of the surrounding tissue. For this reason, vascular development and angiogenesis is only completed once the surrounding tissue stops growing. In a mature or adult organism, vessels acquire a quiescent or dormant state, but they still retain important homeostatic functions and can be reactivated under certain physiological or pathological stimuli. Given their ubiquitous organ distribution and their general relevance for tissue function, growth, healing and regeneration, blood vessels play a very important role in the pathogenesis of most diseases [3]. Research in the last decades revealed the existence of several molecular and cellular mechanisms involved in the regulation of angiogenesis and vascular homeostasis. Some of these mechanisms are important for every aspect of endothelial biology, whereas others have more specific or accessory functions. Similarly to vascular endothelial growth factor (VEGF) signalling, the Notch signalling pathway is among the top regulators of vascular biology. Here we provide an updated review on the role of Notch in endothelial biology.
Notch signalling and its cell context dependency
In mammals, there are four distinct Notch receptors (Notch1-4) that can interact with five different ligands: three of the Delta-like ligand (Dll) family (Dll1, Dll3, Dll4) and two of the Jagged family (Jag1 and Jag2). Both ligands and receptors are transmembrane proteins with large extracellular domains formed by epidermal growth factor (EGF)-like repeats, through which they interact with each other. The activation of the pathway is triggered when Notch receptors interact with transmembrane ligands provided by neighbouring cells [4, 5]. This binding promotes two proteolytic cleavage events in the Notch receptor. The first cleavage is performed by ADAM-family metalloproteases. The second cleavage, which is mediated by a γ-secretase, releases the Notch intracellular domain (Notch ICD or NICD) of the receptor. Then, the cleaved NICD translocates to the nucleus, where it binds the transcription factor RBPJk and recruits co-activators such as Mastermind-like (MAML). This transcriptional complex is able to bind several promoters containing Rbpjk binding sites, including canonical Notch target genes, such as Hes/Hey encoding basic helix-loop-helix (bHLH) transcriptional repressors [6]. The complexity in understanding the role of Notch in cell biology stems from the fact that this apparently simple Notch signalling-transcriptional program varies greatly between different cell types, and also within a single-cell type it can vary significantly according to the genomic and transcriptional status of the cell. This is because the effects of this transcriptional machinery are signalling dose-dependent and also because it interacts with a diverse repertoire of existing transcription factors and chromatin states in the cell [7]. Therefore, knowing the transcriptional programs elicited by Notch in blood cells, neurons, or epithelial cells, will not help us to understand how Notch controls the biology of endothelial cells. As a simple example, Notch activation in blood cells promotes Myc expression and cell proliferation, which can lead to leukemia [8], while in endothelial cells Notch overactivation blocks Myc expression and cell proliferation [9]. Another example is that Notch activation in neurons mostly activates the canonical Hes genes, while in endothelial cells it mostly activates the canonical Hey genes.
Within endothelial cells, it is also becoming clear that Notch triggers completely different transcriptional programs in HUVECs vs HUAECs cultured in vitro [10]. In vivo there are also significant differences between the genetic programs controlled by Notch in growing coronary [9] and retina [11] vessels and even within the same growing retina vascular network, Notch has spatial and temporal-context-dependent roles on EC proliferation [11]. Future single-cell -omics studies will likely expand on this complexity and diversity of Notch functions in single endothelial cells of different organs during development and disease.
Functional relevance of distinct Notch signalling members
Most endothelial cells express mainly the receptors Notch1 and Notch4, and the ligands Dll4 and Jag1, even though all other receptors and ligands are also expressed in endothelial cells [12,13,14,15]. Targeted disruption of different Notch components leads to early embryonic lethality due to severe vascular development defects. Dll4 has been shown to be the most important Notch ligand for early vascular development and angiogenesis. Like the VEGF ligand, its full dose is needed for vascular development. Most mouse embryos lacking a single Dll4 allele are haploinsufficient [16, 17] and die in utero between E9.5-E10.5 with vascular development defects similar to those in Dll4KO embryos. Full loss-of-function mutants for Jag1 [18] and Dll1 [19] show significantly milder vascular development defects, and die at E10.5 and E12, respectively. On the other hand, Jag2 mutants die at birth, but this is due to craniofacial morphogenesis defects [20]. These studies revealed the relative importance of the different ligands for embryonic angiogenesis, but we still do not know well how the different ligands compensate for each other and how they regulate Notch signalling in other organ vascular beds, in adult blood vessels, or in disease. This is particularly relevant since there are now blocking antibodies against each of these ligands [21,22,23,24], and a better understanding of their different functions in physiology and disease could open new therapeutic options.
Similarly to the ligands, we also have now blocking antibodies against the different Notch receptors [25, 26] but we still know little about the functions of the different Notch receptors in endothelial biology, with the exception of studies in early mouse embryos. These studies clearly indicated that Notch1 is the most important receptor for early angiogenesis, since Notch1KO or Notch1floxed−Tie2−Cre embryos die at E9.5 with severe vascular development defects, similarly to RbpjKO embryos [27,28,29]. However, it was also shown that at least Notch4 can partially compensate for Notch1 loss [28], and perhaps Notch2 and Notch3, which are weakly expressed in endothelial cells, may also have an important role in endothelial cells when Notch1 and Notch4 functions are lost. It is interesting to note that most studies in the field have used Rbpjfloxed mice to understand the consequence of disrupting all Notch receptor signalling [17, 30,31,32], but Rbpj is not a transcription factor specific to Notch signalling and may also have Notch-independent roles. In fact, several studies have revealed significant differences between Rbpj and Notch loss-of-function studies in cardiomyocytes, pericytes, and in vitro mouse myogenic cells [33,34,35].
In endothelial cells, it is currently believed that loss of Rbpj generally phenocopies the use of γ-secretase inhibitors or Notch1/Dll4 loss-of-function [11, 16, 17, 21, 22, 28, 36,37,38,39,40] but a thorough phenotypic and RNAseq comparison is needed to fully determine this. The existent higher resolution loss-of-function retina angiogenesis data suggest that there are significant phenotypic differences between Dll4/Notch1/Rbpj mutants [11, 36, 40] being that the increase in vascular density and lack of arterialization after Dll4 deletion is more pronounced than after Notch1 or Rbpj deletion. However, these differences may be because it is easier to delete Dll4 than Notch1 and Rbpj in retina vessels, or the faster turnover of Dll4 in comparison to Rbpj protein. So far not a single study compared the simultaneous deletion of the four Notch receptors to the deletion of Rbpj, something difficult to achieve technically. It has been much easier to combine and induce the deletion of two Rbpj floxed alleles, than of eight floxed alleles corresponding to the four Notch receptors, especially because two of them are located in the same chromosome (Notch3 and Notch4) and thus cannot be combined. The alternative of comparing the effects of Rbpj deletion with general γ-secretase inhibitors (to block all Notch receptors) is also not ideal, since the former represents full genetic deletion, and the latter an incomplete loss of Notch signalling. Our current assumption that Rbpj deletion mimics the loss of all Notch signalling in endothelial cells may be wrong in many cases and should be further tested. We will also need to take this into account if we intend to use RbpjiLOF data to predict the consequence of Notch blockers/inhibitors in the clinics.
Role of Notch in arterial specification
After vasculogenesis, the first and primitive capillary plexus is remodelled into a hierarchical network of vessels containing arteries and veins. Arteries are specialized in delivering oxygenated blood to capillaries, that passively distribute it to surrounding organ cells, and veins collect the low oxygen blood, return it to the heart and close the circuit. For a long time, blood flow dynamics was considered to be the only factor driving arterial-venous specification until genes with an arterial-venous-enriched expression pattern started to emerge [41]. Mature arteries experience high pulsatile shear stress, high blood flow and contain several circumferential and thick layers of smooth muscle cells (SMCs) together with extracellular matrix. On the other hand, veins experience low shear stress, are covered by significantly less SMCs and also have valves to ensure unidirectionality of blood flow [42].
Several studies have shown that the first steps of arterial-venous differentiation occur before significant differences in blood flow are experienced by ECs and the current consensus is that ECs become pre-specified to an arterial or venous fate before artery or vein assembly, even though this process can also be partially modulated by flow. VEGF has been shown to be required for the development of arteries through the activation of Dll4/Notch signalling [43, 44]. Importantly, the expression of Notch ligands and receptors is high in arteries and almost absent in veins [14, 45,46,47].
Genetic deletion of Dll4 or Notch1 is still compatible with the initial development of the first arteries (dorsal aorta) but leads to loss of arterial markers (such as those encoded by Efnb2, Gja4(Cx37) and Gja5(Cx40)) and widespread expression of venous-related genes [16, 17, 48, 49]. The underdeveloped mutant “arteries” have relatively high expression of venous-enriched genes (such as EphB4 and Flt4). Because of this, it was assumed that the default vascular fate was venous, and that high VEGF/Notch signalling was needed to induce an arterial genetic programme on these initially “venous” cells. Questioning the default venous fate, was the finding that COUP-TFII is a vein-specific nuclear receptor that was shown to repress Notch during venous specification. Absence of this transcription factor leads to the acquisition of arterial identity in venous vessels [50]. More recently, COUP-TFII overexpression and single-cell RNA sequencing (scRNAseq) studies in coronary vessels showed again how the ectopic expression of this factor blocks arterialization [47]. Importantly, and in contrast to previous studies, the authors did not see any significant effect of CoupTFIIOE on the expression or activity of Notch receptors. Instead, CoupTFIIOE upregulated the expression of cell cycle genes. In addition, ectopic Notch activation in CoupTFIIOE cells did not rescue their inability to become arterial. These results suggest that modulation of the cell cycle, not Notch, is the key for the effects of CoupTFII on venous development (Fig. 1).

Notch signalling regulates arterial specification by inhibiting endothelial cell cycle genes and biosynthetic activities. The formation of arteries and veins was thought to occur by the induction of a highly conserved arterial and venous genetic programme in a subset of vessels. This specific genetic programme was driven by Notch in arteries and COUP-TFII in veins. Contrary to this established model, recent findings have suggested that arterial-venous pre-specification is tightly linked to the modulation of the endothelial cell cycle and this also influences endothelial cell migration/mobilization towards arterial or venous vessels. Therefore, the formation of arteries does not rely on the direct induction of a Notch-dependent arterial differentiation programme, but instead depends on the suppression of endothelial cell cycle and metabolism, specifically achieved by Myc downregulation
If loss of Notch impairs arterialization, it is expected that ectopic activation of Notch signalling induces arterialization. Indeed, it was shown that during vascular development Notch can repress the expression of venous markers in veins [51,52,53,54]. However, higher resolution mosaic analysis in coronary and retina veins showed that despite the fact that cells with higher Notch signaling have a lower tendency to form veins, they do differentiate normally, losing the expression of arterial marker genes (Cx40) and expressing normal levels of EphB4 and CoupTFII [9]. Only in pre-arterial capillaries, cells with high Notch signalling had higher expression of arterial marker genes. In pre-venous or venous vessels, these cells had normal expression of venous marker genes. This study also showed that cells with high Notch proliferated significantly less, but they differentiated adequately to venous cells when located in the right venous microenvironment (Fig. 1).
Even though there are several arterial enriched genes, few have such an important role as Notch in arterialization, and hence it is considered a master regulator of arterialization. For example, Cx37 and Cx40 are excellent markers of the arterial phenotype, and strongly activated by Notch, but their dual loss do not affect embryonic arterialization. Notch also strongly regulates the expression of the arterial-specific Ephrin-B2 ligand, and knockout of this ligand also results in arterial development defects [55].
Besides the requirement of Notch for the development of the dorsal aorta in mouse and zebrafish embryos, its relevance for the development of other arteries has also been demonstrated more recently by genetic deletion of Notch signalling components in developing coronary, retina and brain vessels. The complete loss of Dll4-Notch signalling also significantly compromises arterial development in these organs [36, 40, 56,57,58]. This data suggests that most organ arteries may require Notch signalling in order to develop. Indeed, a recent study using Apelin-driven RbpjKO genetic mosaics showed that these cells poorly contribute to the formation of adult aorta, liver, brain, heart and retina arteries [9]. However, in these mosaic mutants a significant fraction of RbpjKO cells can still form arteries, which raises questions about the essential cell-autonomous function of Notch in arterialization.
Recent publications using pulse-chase lineage tracing genetics in mice or live imaging in zebrafish have provided significantly more insight into the process of arterial specification, and how it involves not only genetic pre-determination but also mobilization. These studies have shown that vein-derived ECs and tip cells with high CXCR4/Notch signalling migrate against the flow to be incorporated into nascent arteries [59,60,61]. The concept of arterial development accomplished by migratory cells is supported by the phenotype resulting from deletion of Cdc42 during retina vascular growth. Absence of this small GTPase inhibits EC migration and leads to EC accumulation in venous areas [62]. In addition, recent scRNAseq studies with developing coronary ECs collected at different stages showed that there is a continuum of EC identities along the arterial-venous axis [47]. A subset of endothelial cells go through a gradual switch from a venous-like programme to a pre-arterial one, well before the main coronary arteries form and there is pulsatile flow. Lineage tracing revealed that ECs with high CXCR4, CX40 and Notch signalling are more committed/biased to mobilize and form arteries. On the contrary, the vein-determining transcription factor COUP-TFII suppresses pre-arterial transition by activating cell cycle genes [47]. This close relationship between veins and cell proliferation and between arrested tip cells and arteries seems to be general and is also supported by other single-cell transcriptomic and epigenomic studies [63]. In line with these results, we have shown recently that it is not the Notch-dependent arterial differentiation program, but instead the ability of Notch to suppress Myc-dependent endothelial metabolism and cell cycle progression, which drives arterial mobilization and development [9]. Using standard conditional genetics and genetic mosaics we have shown that RbpjKO cells rarely form arteries, however Rbpj/MycDKO cells do so equally efficiently as wildtype cells. These results show that Notch/Rbpj signalling does not directly induce arterial specification, since it can occur in its absence. Indeed, ectopic activation of Notch in different endothelial cell lines is not sufficient to arterialize cells [10], even though it biases the cells towards arterial mobilization in vivo [9]. Since Myc mainly controls cell metabolism and growth, and not directly cell motility, this suggests that inhibition of the cell cycle or metabolism by the Notch-dependent suppression of Myc may render cells more permissive for pre-arterial specification or more chemotactic towards arteries. In line with this, very high VEGF activity, induced in a mosaic and cell-autonomous manner, also decreases EC proliferation and increases the cell´s ability to mobilize and form arteries. Most retina Esm1 + tip cells are also positive for the cyclin-dependent kinase inhibitor (p21), and proliferate significantly less than stalk cells [11], and these cells were also shown to effectively mobilize to arteries [59,60,61]. Finally, the use of cell cycle inhibitors during vascular development also enhances arterial specification [47]. These studies indicate that arterial-venous pre-specification is tightly linked to the modulation of the endothelial cell cycle, which also influences EC migration/mobilization towards arterial or venous vessels (Fig. 1). During tissue development, this genetic or cell cycle/metabolic specification occurs before significant differences in blood flow exist. Nonetheless, blood flow and shear stress may help later to reinforce these pre-existing genetic programs by providing additional biophysical factors that further enhance the arteriovenous differences. Indeed, Notch itself is a mechanosensitive pathway and arterial shear stress is able to enhance Notch signalling. This has been also shown to supress endothelial proliferation and trigger the expression of an arterial-specific program [64,65,66].
A better understanding of how all these biophysical and genetic factors are spatially and temporally integrated in vivo is not only important to gain more knowledge, but also because it may allow the development of better therapeutic strategies to induce effective arterialization after tissue ischemia, such as after coronary obstruction and myocardial infarction.
Regulation of sprouting angiogenesis by Notch signalling
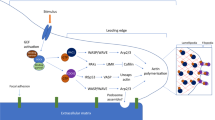
The growth of the vascular network is an extremely dynamic process and requires multiple and coordinated steps that include EC proliferation, sprouting and migration. When a tissue does not have enough oxygen, it secretes pro-angiogenic factors such as VEGF that, by binding to its receptors, activates a fraction of ECs and confer these a motile and invasive phenotype [67,68,69]. These so-called tip cells acquire long filopodia and lead the sprouting process. Following these invasive and migratory cells, are the stalk cells, the building blocks for the budding sprout. These cells establish a vascular lumen for blood flow and also actively proliferate to sustain sprout elongation [70, 71] Most of the research done in mice to reveal tip/stalk cell biology has been performed in the postnatal mouse retinal angiogenesis model, which is a relatively easy system to genetically manipulate and image [72]. Using this system it has been shown that the biology of tip and stalk cells is strongly regulated by the VEGF/Notch pathway [69, 73]. VEGF is one of the most potent pro-angiogenic factors and its blockade results in severe retinal angiogenesis defects, with a marked decrease in the number of sprouting cells [40, 74]. VEGF gradient and concentration finely regulate tip cell migration and stalk cell proliferation [67]. Mechanistically, VEGF-induced VEGFR2 signalling triggers several phosphorylation cascades, including the downstream mitogen-activated protein kinase (MAPK)/ extracellular signal regulated kinase (ERK) signalling activation [75, 76]. In addition, VEGFR2 signalling leads to the transcription of tip cell-enriched genes such as Esm1 and Dll4. Tip cells that express more Dll4, in turn activate Notch signalling in the adjacent cells. Consequently, this maintains the stalk fate in adjacent cells by suppressing tip cell behaviour [21, 22, 36, 37]. Deletion of just one Dll4 allele is sufficient to induce excessive sprouting and vessel branching because of increased tip cell formation. This is accompanied by an increase in tip cell-enriched genes such as PdgfB, Unc5b [38], Esm1, or Apln [77] in the rest of the vasculature. Loss of Notch signalling by deleting Dll4, Notch1 or Rbpj leads to pronounced vascular developmental defects that were initially related with a general increase in EC activity and angiogenesis [16, 21, 22, 36,37,38,39]. Conversely, Notch gain-of-function studies showed that it decreases vascular density and reduced tip cell number and EC sprouting [38, 78, 79]. Manipulation of Notch signalling activity in a mosaic manner confirmed the role of Notch signalling in tip cell specification [36, 75]. Live imaging and fate-mapping studies in zebrafish and mouse models have shown that tip cell specification is likely transient and oscillatory, since ECs can switch between stalk and tip positions during angiogenesis. This dynamic process is considered to be regulated by the integration of VEGF and Notch signalling levels, which drive a continuous shuffling of tip and stalk cells competing for the lead position [80, 81]. Since in other developmental systems Notch has been shown to be activated in an oscillatory manner [82], oscillations in the levels of Notch signalling may be also involved in the regulation of the observed oscillatory tip/stalk cell fates and dynamics. In addition, signaling through bone morphogenetic proteins and SMAD1/5 [83,84,85], and remodelling of VE-cadherin junctions [86] have also been shown to be involved in tip/stalk cell interconversion, and these molecular mechanisms can also be regulated upstream by Notch/Heys and VEGF signalling. Furthermore, ECs, and particularly tip cells, have been shown to polarize against blood flow and form the source for arterial development, both in the mouse retina and in zebrafish [59, 60, 87]. Nevertheless, only a fraction of them seem to be able to do so, as most Esm1 + tip cells pulse-chased at postnatal day 3 (P3) have not incorporated into arteries at P6 [11] (Fig. 2).

source and lower Notch activity. Tip cell specification has been shown to be transient and oscillatory. ECs at the leading edge are exposed to varying growth and biophysical factors, which makes them having different abilities to compete for the tip position. This continuous sensing and reaction to the surrounding tissue microenvironment results in a dynamic exchange among tip and stalk cells during angiogenesis.
ECs have a bell-shaped dose-dependent response to mitogenic stimuli in vivo. By binding to its cognate receptor VEGFR2, VEGF activates the fraction of ECs that become leading tip cells, triggering several downstream cascades, including the MAPK/ERK signalling pathway. These high VEGF/ERK signalling cells also express p21, which results in an arrested and non-proliferative EC phenotype. VEGFR2 signalling also results in Dll4 expression on tip cells, which in turns activates Notch signalling in the adjacent stalk cells and suppresses the tip cell phenotype by downregulating ERK signalling. Therefore, physiological Notch signalling in stalk cells is essential to prevent the hypermitogenic arrest of these ECs and sustain angiogenesis. Overall, recent studies have shown that adequate VEGF and Notch signalling balance must be maintained in order for ECs to proliferate and sprout/migrate adequately. Jagged1 is a pro-angiogenic regulator mainly expressed in stalk cells which antagonizes Dll4-Notch signalling from stalk cells to tip cells. Contrary to previous studies, it has been shown that tip cells express similar amounts of VEGFR2 protein as stalk cells but they have higher ERK activity, likely because of their high proximity to the VEGF
The potent effect of Dll4/Notch as a suppressor of sprouting angiogenesis was also shown in pathological conditions. Dll4/Notch inhibition resulted in a significant decrease in tumour growth due to excessive but non-productive sprouting angiogenesis [21, 22]. The paradoxal reduction in tumour growth by Notch blockade, even after triggering an increase in sprouting angiogenesis, was explained by the authors as a consequence of a decrease in tumour blood perfusion when vessels are excessively branched.
In addition to Dll4, another Notch ligand was found to have an important and opposing role in tip/stalk cell biology and angiogenesis. Jagged1 was found to be a pro-angiogenic regulator mainly expressed in stalk cells which antagonizes Dll4-Notch signalling in Fringe glycosyltransferases (Mfng/Lfng) expressing ECs, including tip cells [39]. By glycosylating the Notch receptors, Fringes polarize the function of the Delta and Jagged ligands [88, 89] in ECs. In the presence of glycosylated Notch receptors, Delta becomes a stronger ligand while Jagged becomes a weaker ligand, but since both bind the same receptors, Jagged1 reduces endothelial Dll4/Notch signalling. Therefore, the relative expression of and equilibrium between different Notch ligands and modulators control and reinforce the different behaviours between endothelial tip cells (Notch signalling sending) and stalk cells (Notch signalling receiving cells) (Fig. 2).
For many years, the VEGF/Notch interplay in tip/stalk cell differentiation was thought to rely on the suppression of VEGFR2 transcription by Dll4-Notch activation in stalk cells, which was shown to happen in cultured HUVECs [69, 90, 91]. However, several other studies showed that Notch does not significantly regulate the transcription of Vegfr2 in vivo in retina, heart and zebrafish vessels [9, 11, 37, 40, 92] and that tip cells express similar amounts of Vegfr2 protein as stalk cells [40]. These studies also showed that unlike Vegfr2, Vegfr3 transcription and protein levels are more tightly regulated by Notch signalling in vivo, however this receptor does not bind VEGF-A (only VEGF-C/D) and its role is more important for lymphangiogenesis [42], even though it can also heterodimerize with VEGFR2 and potentially regulate its function [74]. Nonetheless, all available evidence shows that tip cells have higher MAPK/ERK activity, which is strongly activated by VEGF signalling and suppressed by Notch signalling [11, 76]. Tip cells have relatively higher ERK activity, likely because of the combination of two factors, being the higher proximity to the VEGF source, and the lower Notch signalling activity. The latter could be due to Dll4 cis-Notch signalling inhibition in tip cells plus the weaker expression of Dll4 (and stronger Jagged1) ligands by stalk cells. Demonstrating the key role of ERK activity in tip cell biology, suppression of MEK/ERK signalling with pharmacological compounds completely prevents EC sprouting or migration in zebrafish and in the retinal angiogenesis model [11, 76].
Given that tip cells have higher MEK/ERK activity, one would expected these to proliferate more than stalk cells. However, in vivo studies have shown that retina tip cells, particularly VEGF/Esm1-high tip cells, are less proliferative than their neighbouring stalk cells [11, 67]. A recent study has shown that ECs have a bell-shaped dose-dependent response to mitogenic stimuli in vivo [11]. When mitogenic stimulation is increased by Dll4/Notch signalling inhibition, or by VEGFR2 signalling overactivation, the most angiogenic and proliferative ECs located at the angiogenic front exit the cell cycle. Although in the first 24 h ECs subjected to loss of Notch signalling experience a transient increase in the speed of cell cycle progression and division, this is followed by a pronounced cell cycle arrest at 48 h after the start of the inhibition. This arrest is partially mediated by the strong upregulation of ERK activity and p21. Therefore, even though Notch overactivation does block endothelial cell proliferation, physiological Notch signalling does not inhibit angiogenesis but is instead essential to prevent the arrest of endothelial stalk cells at the angiogenic vascular front, thus sustaining angiogenesis [11]. During physiological retinal angiogenesis, higher ERK signalling and p21 expression (inducing cell cycle arrest) is restricted to tip ECs, being the cells with lower Notch signalling and higher VEGF signalling. The cell cycle arrest of tip cells is likely necessary to enhance their sprouting/migration ability. These studies also show that an adequate VEGF and Notch signalling balance must be maintained in order for ECs to proliferate and sprout/migrate adequately. These results may also partially explain the failure of therapeutic pro-angiogenic strategies, such as the delivery of growth factors after myocardial infarction [93, 94]. This simple approach is likely not enough to effectively stimulate angiogenesis since vessels elicit cell cycle checkpoint mechanisms at high levels of growth factor stimulation. Understanding these hypermitogenic arrest mechanisms in more detail may shed more light on ways of effectively boosting therapeutic angiogenesis (Fig. 2).
Role of Notch in vascular maturation and quiescence
After the angiogenic expansion of the blood vessel network, vessels undergo maturation in order to form a functional vascular system. This maturation includes vascular pruning, remodelling, stabilization and finally cellular differentiation, and culminates with the acquisition of a quiescent state [42, 71]. During the process of vascular maturation, there is a progressive switch from active growth to a quiescent state. This switch relies mostly on the withdrawal of pro-angiogenic microenvironmental cues but also cell-intrinsic and cell-to-cell contact molecular mechanisms [95]. This leads to a significant decrease in endothelial proliferation and the markers of angiogenic activity or sprouting. Most quiescent ECs do not proliferate and have a very low metabolic and gene expression profile. Quiescence is however a reversible state. The quiescent phenotype is in general maintained until ECs detect pro-angiogenic signals that can rapidly switch them from a long-term quiescence to an active growth state [71]. In an adult organism, the maintenance of a quiescent and functional vascular network is critical for the structure and function of organ parenchyma, maintaining tissue homeostasis. It is also becoming clear that maintenance of vascular quiescence and homeostasis is not a passive process, but involves the activity of several signalling pathways, most of which were already introduced before as being key regulators of angiogenesis.
Many signalling pathways required for the development of blood vessels have been associated with the vascular maturation process, and the recruitment of endothelial supporting cells, including pericytes and vascular smooth muscle cells (VSMCs), has been shown to stabilize nascent blood vessels and regulate the secretion of important extracellular matrix components [67, 96, 97].
Besides the essential role of Notch during vasculogenesis and angiogenesis, it has been shown that Notch signalling it is also required throughout the entire vascular maturation process. In the mouse retina model, it has been shown that Dll4-Notch signalling activity is essential for the pruning and maturation of veins and perivenous capillaries, but surprisingly not for the maintenance of arteries [78]. Endothelial-specific deletion of Rbpj leads to reduced vessel pruning and mural cell recruitment, with an increase in vessel sprouting and proliferation only in the venous areas. Despite the Notch pathway components being highly expressed in the arterial endothelium throughout life, they do not seem to have a major function in their homeostasis [78]. Loss of Notch1 in aorta endothelial cells results in a very mild increase in EC proliferation [65]. In contrast to angiogenic ECs, loss of Dll4/Notch signalling in the maturing vascular plexus of the retina leads to cell cycle reentry [78], instead of cell cycle exit, which is likely due to the lower basal levels of VEGF/mitogenic stimuli in maturing/remodelling vessels and the identified bell-shaped dose-dependent response to mitogenic stimulation [11]. This highlights once again how much the role of Notch and VEGF in EC biology depend on mitogenic context.
Several studies also showed that the Notch signalling pathway is essential to maintain the quiescence of some organ vessels and their homeostasis. Pharmacological inhibition or genetic deletion of different Notch components such as Dll4, Notch1, or Rbpj in the adult and fully mature endothelium results in enlarged vessels and increased vascular density and in some studies this was linked to an increase in EC proliferation and vascular neoplasms [15, 98,99,100,101]. Overall, these studies raised important concerns against the use of Notch inhibitors in the clinics and questioned the safety of targeting the Dll4-Notch1 signalling axis. Targeting this signalling pathway was before seen as a very promising approach against cancer, since Dll4 blockade with Dll4-Fc or anti-Dll4 monoclonal antibodies significantly reduced tumour growth, even in tumours resistant to anti-VEGF treatment [21, 22]. Now, these more recent studies on the effect of Notch blockers in vascular quiescence reveal potential secondary effects of those treatments and raise a red flag in the use of anti-Dll4/Notch for cancer treatment. Other studies suggested that the anti-Dll4/Notch effects on quiescent vessels are reversible after treatment withdrawal and may have less impact on organ physiology than initially proposed [102].
The mechanisms triggered by Notch activation that induce EC quiescence are still incompletely understood. Mechanisms identified by overactivation of Notch signalling in cultured cells may also be misleading, since the range of genetic targets activated by Notch signalling is dose-dependent and not easily modelled in gain-of-function assays, particularly in vitro. Nonetheless, several factors downstream of Notch have been suggested to be responsible for inducing EC quiescence by repressing proliferation both in vitro and in vivo, such as Interleukin-33, p27, thrombospondin-1, p21 and PTEN [103,104,105,106]. Altogether, these studies point to Notch activation being a pro-quiescence pathway that is essential to maintain vascular homeostasis [15, 98,99,100,101].
Some other studies have linked the role of Notch in vascular quiescence with shear stress. These studies suggested that vascular quiescence is a blood flow or shear stress-dependent process, which is mediated by Notch-mediated suppression of the cell cycle and the stabilization of cell-to-cell contact mechanisms. These studies proposed that Notch signalling activity is regulated by shear stress not only during arterial development but also during adult arterial homeostasis [65, 66]. Surprisingly, a non-canonical and transcriptional-independent Notch-mediated mechanism was also proposed to be involved in endothelial barrier maintenance by Notch. Shear stress promotes Dll4-dependent proteolytic cleavage of the Notch transmembrane domain, which increases its availability to complex with other proteins as VE-cadherin, and the Rac1 guanidine-exchange factor TRIO. This complex allows to control adherens junctions and barrier function independently of the role of Notch signaling in transcriptional regulation [107].
Role of Notch in cardiovascular disease
The role of Notch in the adult vasculature after induced cardiovascular disease was also analysed. In the hindlimb ischemia model, Dll1 haploinsufficieny impairs arterial remodelling and blood flow restoration. Dll1 expression is restricted to arterial endothelium and its upregulation seems to be required for the Notch-dependent arteriogenesis [108]. In line with this, Notch1 and Notch4 have also been shown to be required for proper angiogenesis and blood flow recovery in hindlimb ischemia models [109]. In another study, Dll4 heterozygous mice were found to have more pial collateral arteries and have reduced blood flow conductance after femoral artery occlusion. Despite markedly increased angiogenesis, tissue ischemia was more severe in these mice [110].
The role of Notch signalling in atherosclerosis has also been evaluated. Some reports have shown that endothelial deletion of Rbpj reduces atherosclerosis progression, with decreased secretion of inflammatory factors and leukocyte recruitment [111]. In contrast, others studies have suggested an atheroprotective role for Notch signalling and increased expression of pro-inflammatory genes in the absence of Notch1 [65]. Despite these opposing results, and diversity of the proposed mechanisms, endothelial Notch signalling seems to be very relevant for cardiovascular disease and future studies will refine its role in these processes.
Angiocrine functions of endothelial Notch signalling
Apart from the widely known function of blood vessels in delivering oxygen and nutrients, ECs are also involved in the control of an extensive variety of other physiological processes. Some of these important EC roles rely on the expression and secretion of either stimulatory or inhibitory growth factors, morphogens or chemokines, popularly referred as angiocrine factors. It has been shown that these endothelial secreted factors are able to regulate the homeostasis, self-renewal, growth and differentiation of neighboring cells or resident stem cells [112]. Although Aristotle already suggested an essential role of vascular structures during organogenesis [113], it was shown approximately 20 years ago that liver sinusoidal ECs were involved in liver bud development in a perfusion-independent manner [114]. Since then, many studies have listed genes expressed by ECs, which can code for potential angiocrine factors. Among those signals, Notch was proposed to be such an angiocrine factor for its ability to transmit signals by cell-to-cell contact.
One of the first results showing an angiocrine role for endothelial Notch was obtained by analysing the role of the Notch pathway in the bone marrow endothelium and associated hematopoiesis. It was found that upon myeloablation, ECs from the bone marrow niche are able to support the self-renewal of the hematopoietic stem cell (HSC) pool through expression of the Jagged1 and Jagged2 ligands and subsequent Notch activation in HSCs [115, 116]. Later it was also found that in steady-state conditions, endothelial expression of Dll1 in the bone marrow and spleen regulates monocyte fate, favouring the conversion of Ly6Chi monocytes into Ly6Clow monocytes. However, during ischemia, Dll1 upregulation in arterial ECs leads instead to differentiation of Ly6Chi monocytes into macrophages that promote arteriogenesis and allow tissue repair [117, 118]. It was also found in the bone marrow that endothelial Notch signalling induces the expression and secretion of the angiocrine factor Noggin. This bone morphogenic protein is required for chondrocyte and osteoblast homeostasis [119].
Notch activation can also be detrimental for the homeostasis or biology of the surrounding tissue. Notch activation in sinusoidal liver ECs downregulates important hepatocyte mitogens such as Wnt2, Wnt9b and hepatocyte growth factor [120]. This leads to impaired hepatocyte proliferation not only during organ homeostasis but also in regenerating conditions.
It was also shown in liver that mechanically stretched sinusoidal ECs upregulate Cxcl1 via Notch. The secretion of this angiocrine factor leads to neutrophil recruitment and subsequent formation of neutrophil extracellular traps with microthrombi [121]. Moreover, it has been shown that high levels of VEGFR2/Notch signalling in ECs results in myocardial hypertrophy. An increase of endothelial VEGF/VEGR2 signalling leads to Notch activation in ECs, which results in increased expression of Adam12 and Klk8. The upregulation of these two genes allows the release of the heparin-binding-EGF and neuregulin1 ligands by ECs, which promote their binding to ErbB receptors in neighbouring cardiomyocytes, and consequently enhance their growth [122]. Repetitive lung injury was also shown to result in the upregulation of Jag1 by endothelial cells. This ligand activates Notch signalling in perivascular fibroblasts and consequently enhances fibrosis [123].
A comprehensive knowledge of the organ-specific angiocrine functions by Notch is required to understand and predict the impact of pharmacological targeting of this pathway in translational vascular medicine. The promising therapeutic potential of tissue-specific and resident ECs may represent a target for angiocrine-mediated tissue regeneration/biology, or for the amelioration of certain conditions such as fibrosis [112].
Role of Notch on vascular heterogeneity arising from single-cell studies
Recent scRNA-seq studies have allowed the comparative analysis of the transcriptome of most ECs within most adult organ vascular beds [63, 124,125,126,127,128,129,130,131,132,133].
Bioinformatics analyses of single-cell experiments were used to study the differentiation of different endothelial cell types during vascular development. An elegant scRNA-seq study on developing heart vessels showed that venous cells go through a gradual switch from a venous programme to an arterial one characterized by higher Notch activity and expression of target genes (Efnb2, Hes1, Hey2, Gja5 and Gja4) during heart development [47]. Analysis of adult organ vessels has also revealed that there is a continuum of Notch-related single endothelial cell identities along the arterial-venous axis [14, 63, 133].
In the liver it was also found that this arterial-venous vascular continuum correlated with the surrounding tissue spatial heterogeneity or zonation. By combining single-molecule in situ hybridization, scRNA-seq and paired-cell sequencing, recent studies spatially reconstructed the endothelial-hepatocyte zonation [134, 14]. Interestingly, the spatial zonation related with Notch signalling follows an oxygen gradient. Notch components and target genes are highly expressed in the oxygenated portal nodes, which comprise the hepatic arteries and portal veins. Notch expression and activity progressively decreases continuously in capillaries towards the hepatic central veins [14]. Given the role of Notch as a master regulator of cellular and transcriptional heterogeneity it will be interesting to know its role in the specification of this arterial-venous diversity at the single-cell level in different adult organ vascular beds.
Conclusions
In the last years there has been a substantial progress in the understanding of Notch signalling in endothelial cell proliferation, sprouting, migration, vessel stabilization and maturation. Both in vitro and in vivo studies have shown how the spatial and temporal-context of an endothelial cell, strongly influences the role of Notch on its specification, proliferation and homeostasis. Dispite significant progress, still little is known about the complex genetic programmes triggered by Notch that support these context-dependent differences. Future single-cell and genetic studies and related breakthrough technologies will certainly expand our understanding of how Notch controls endothelial heterogeneity in different organs and tissues during development, homeostasis and disease. The challenge will be to integrate the immense datasets that will be generated and functionally validate the therapeutic relevance of those findings.
While the induction of therapeutic angiogenesis may be advantageous in cardiovascular diseases such as heart and brain ischemia, hypovascular-related dementia, neurodegeneration and wound healing, its inhibition would be beneficial in inflammatory disorders, diabetes and cancer [1, 3]. Even though targeting Notch signalling as a whole is not as vascular-specific as targeting VEGF signalling, it is still one of the most important pathways for vascular biology and homeostasis. In a adult organism, Dll4 (and Notch4) is mainly expressed by ECs, and therefore it is still an attractive vascular-specific therapeutic target, particularly if issues related with the induction of vascular neoplasms after its inhibition in quiescent vessels are resolved. Future studies may identify ways to interfere specifically with the Dll4/Notch function during angiogenesis without compromising its protective role on vascular homeostasis.
Therefore, a better understanding of the mechanisms that control angiogenesis versus vascular homeostasis may be of high therapeutic relevance. This may lead to the identification of treatments with higher biomedical precision and less undesired side effects.
References
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936
Risau W, Flamme I (1995) Vasculogenesis. Annu Rev Cell Dev Biol 11:73–91
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473:298–307
Andersson ER, Sandberg R, Lendahl U (2011) Notch signaling: simplicity in design, versatility in function. Development 138:3593–3612
Kopan R, Ilagan MX (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137:216–233
Hori K, Sen A, Artavanis-Tsakonas S (2013) Notch signaling at a glance. J Cell Sci 126:2135–2140
Bray SJ (2006) Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7:678–689
Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O’Neil J, Neuberg D, Weng AP et al (2006) NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci 103(48):18261–18266. https://doi.org/10.1073/pnas.0606108103
Luo W, Garcia-Gonzalez I, Fernández-Chacón M, Casquero-Garcia V, Sanchez-Muñoz MS, Mühleder S, Garcia-Ortega L, Andrade J, Potente M, Benedito R (2021) Arterialization requires the timely suppression of cell growth. Nature 589(7842):437–441
Aranguren XL, Agirre X, Beerens M, Coppiello G, Uriz M, Vandersmissen I, Benkheil M, Panadero J, Aguado N, Pascual-Montano A, Segura V, Prósper F, Luttun A (2013) Unraveling a novel transcription factor code determining the human arterial-specific endothelial cell signature. Blood 122(24):3982–3992
Pontes-Quero S, Fernandez-Chacon M, Luo W, Lunella FF, Casquero-Garcia V, Garcia-Gonzalez I, Hermoso A, Rocha SF, Bansal M, Benedito R (2019) High mitogenic stimulation arrests angiogenesis. Nat Commun 10:2016
Hofmann JJ, Iruela-Arispe ML (2007) Notch signaling in blood vessels: who is talking to whom about what? Circ Res 100(11):1556–1568
Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, Ding BS, Schachterle W, Liu Y, Rosenwaks Z et al (2013) Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell 26:204–219
Halpern KB, Shenhav R, Massalha H, Toth B, Egozi A, Massasa EE, Medgalia C, David E, Giladi A, Moor AE et al (2018) Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat Biotechnol 36:962–970
Jabs M, Rose AJ, Lehmann LH, Taylor J, Moll I, Sijmonsma TP, Herberich SE, Sauer SW, Poschet G, Federico G et al (2018) Inhibition of endothelial Notch signaling impairs fatty acid transport and leads to metabolic and vascular remodeling of the adult heart. Circulation 137:2592–2608
Duarte A, Hirashima M, Benedito R, Trindade A, Diniz P, Bekman E, Costa L, Henrique D, Rossant J (2004) Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev 18:2474–2478
Krebs LT, Shutter JR, Tanigaki K, Honjo T, Stark KL, Gridley T (2004) Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev 18:2469–2473
Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, Gendron-Maguire M, Rand EB, Weinmaster G, Gridley T (1999) Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet 8:723–730
Hrabe de Angelis M, McIntyre J 2nd, Gossler A (1997) Maintenance of somite borders in mice requires the Delta homologue DII1. Nature 386:717–721
Jiang R, Lan Y, Chapman HD, Shawber C, Norton CR, Serreze DV, Weinmaster G, Gridley T (1998) Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev 12(7):1046–1057
Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I et al (2006) Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444:1083–1087
Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G (2006) Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444:1032–1037
Chiorean EG, LoRusso P, Strother RM, Diamond JR, Younger A, Messersmith WA, Adriaens L, Liu L, Kao RJ, DiCioccio AT, Kostic A, Leek R, Harris A, Jimeno A (2015) A phase i first-in-human study of enoticumab (REGN421), a fully human delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin Cancer Res 21(12):2695–2703
Lafkas D, Shelton A, Chiu C, de Leon Boenig G, Chen Y, Stawicki SS, Siltanen C, Reichelt M, Zhou M, Wu X, Eastham-Anderson J, Moore H, Roose-Girma M, Chinn Y, Hang JQ, Warming S, Egen J, Lee WP, Austin C, Wu Y, Siebel CW (2015) Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature 528(7580):127–131
Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, Choy R, Howard P, Kadyk L, Yan M, Siebel CW (2010) Therapeutic antibody targeting of individual Notch receptors. Nature 464(7291):1052–1057
Moore G, Annett S, McClements L, Robson T (2020) Top Notch in cancer: a detailed overview of recent insights and current perspectives. Cells 9(6):1503
Oka C, Nakano T, Wakeham A, de la Pompa JL, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak TW, Honjo T (1995) Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development 121(10):3291–3301
Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R et al (2000) Notch signaling is essential for vascular morphogenesis in mice. Genes Dev 14(11):1343–1352
Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK (2005) Essential role of endothelial Notch1 in angiogenesis. Circulation 111:1826–1832
Buono KD, Robinson GW, Martin C, Shi S, Stanley P, Tanigaki K, Honjo T, Hennighausen L (2006) The canonical Notch/RBP-J signaling pathway controls the balance of cell lineages in mammary epithelium during pregnancy. Dev Biol 293(2):565–580
Vasyutina E, Lenhard DC, Wende H, Erdmann B, Epstein JA, Birchmeier C (2007) RBP-J (Rbpsuh) is essential to maintain muscle progenitor cells and to generate satellite cells. Proc Natl Acad Sci 104(11):4443–4448
Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F, Han H, Meng A, Wang Y, Yang X (2011) Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev Cell 20(3):291–302
Castel D, Mourikis P, Bartels SJ, Brinkman AB, Tajbakhsh S, Stunnenberg HG (2013) Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev 27:1059–1071
Diaz-Trelles R, Scimia MC, Bushway P, Tran D, Monosov A, Monosov E, Peterson K, Rentschler S, Cabrales P, Ruiz-Lozano P et al (2016) Notch-independent RBPJ controls angiogenesis in the adult heart. Nat Commun 7:12088
Diéguez-Hurtado R, Kato K, Giaimo BD, Nieminen-Kelhä M, Arf H, Ferrante F, Bartkuhn M, Zimmermann T, Bixel MG, Eilken HM, Adams S, Borggrefe T, Vajkoczy P, Adams RH (2019) Loss of the transcription factor RBPJ induces disease-promoting properties in brain pericytes. Nat Commun 10(1):2817
Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N et al (2007) Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445:776–780
Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ (2007) Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci 104:3219–3224
Suchting S, Freitas C, le Noble F, Benedito R, Breant C, Duarte A, Eichmann A (2007) The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci 104:3225–3230
Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH (2009) The Notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137:1124–1135
Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH (2012) Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 484:110–114
Adams RH (2003) Molecular control of arterial-venous blood vessel identity. J Anat 202:105–112
Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8:464–478
Lawson ND, Vogel AM, Weinstein BM (2002) sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell 3:127–136
Seo S, Kume T (2006) Forkhead transcription factors, Foxc1 and Foxc2, are required for the morphogenesis of the cardiac outflow tract. Dev Biol 296:421–436
Villa N, Walker L, Lindsell CE, Gasson J, Iruela-Arispe ML, Weinmaster G (2001) Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev 108:161–164
Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, Weinstein BM (2001) Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128(19):3675–3683
Su T, Stanley G, Sinha R, D’Amato G, Das S, Rhee S, Chang AH, Poduri A, Raftrey B, Dinh TT et al (2018) Single-cell analysis of early progenitor cells that build coronary arteries. Nature 559:356–362
Gridley T (2010) Notch signaling in the vasculature. Notch signaling. Elsevier, Amsterdam, pp 277–309
Swift MR, Weinstein BM (2009) Arterial-venous specification during development. Circ Res 104(5):576–588
You LR, Lin FJ, Lee CT, Demayo FJ, Tsai MJ, Tsai SY (2005) Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature 435:98–104
Carlson TR, Yan Y, Wu X, Lam MT, Tang GL, Beverly LJ, Messina LM, Capobianco AJ, Werb Z, Wang R (2005) Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc Natl Acad Sci U. S. A 102:9884–9889
Murphy PA, Kim TN, Lu G, Bollen AW, Schaffer CB, Wang RA (2012) Notch4 normalization reduces blood vessel size in arteriovenous malformations. Sci Transl Med 4(117):117ra8
Trindade A, Kumar SR, Scehnet JS, Lopes-da-Costa L, Becker J, Jiang W, Liu R, Gill PS, Duarte A (2008) Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 112(5):1720–1729
Venkatesh DA, Park KS, Harrington A, Miceli-Libby L, Yoon JK, Liaw L (2008) Cardiovascular and hematopoietic defects associated with Notch1 activation in embryonic Tie2-expressing populations. Circ Res 103(4):423–431
Wang HU, Chen Z-F, Anderson DJ (1998) Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93:741–753
Kim YH, Hu H, Guevara-Gallardo S, Lam MT, Fong SY, Wang RA (2008) Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development 135(22):3755–3764
D’Amato G, Luxan G, del Monte-Nieto G, Martinez-Poveda B, Torroja C, Walter W, Bochter MS, Benedito R, Cole S, Martinez F et al (2016) Sequential Notch activation regulates ventricular chamber development. Nat Cell Biol 18:7–20
Travisano SI, Oliveira VL, Prados B, Grego-Bessa J, Pineiro-Sabaris R, Bou V, Gomez MJ, Sanchez-Cabo F, MacGrogan D, de la Pompa JL (2019) Coronary arterial development is regulated by a Dll4-Jag1-EphrinB2 signaling cascade. Elife 8:e49977
Hasan SS, Tsaryk R, Lange M, Wisniewski L, Moore JC, Lawson ND, Wojciechowska K, Schnittler H, Siekmann AF (2017) Endothelial Notch signalling limits angiogenesis via control of artery formation. Nat Cell Biol 19:928–940
Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D, Rocha SF et al (2017) Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol 19:915–927
Xu C, Hasan SS, Schmidt I, Rocha SF, Pitulescu ME, Bussmann J, Meyen D, Raz E, Adams RH, Siekmann AF (2014) Arteries are formed by vein-derived endothelial tip cells. Nat Commun 5:5758
Lavina, B., Castro, M., Niaudet, C., Cruys, B., Alvarez-Aznar, A., Carmeliet, P., Bentley, K., Brakebusch, C., Betsholtz, C., and Gaengel, K. (2018). Defective endothelial cell migration in the absence of Cdc42 leads to capillary-venous malformations. Development 145
Sabbagh MF, Heng JS, Luo C, Castanon RG, Nery JR, Rattner A, Goff LA, Ecker JR, Nathans J (2018) Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. Elife 7:e36187
Gordon WR, Zimmerman B, He L, Miles LJ, Huang J, Tiyanont K, McArthur DG, Aster JC, Perrimon N, Loparo JJ et al (2015) Mechanical allostery: evidence for a force requirement in the proteolytic activation of Notch. Dev Cell 33:729–736
Mack JJ, Mosqueiro TS, Archer BJ, Jones WM, Sunshine H, Faas GC, Briot A, Aragon RL, Su T, Romay MC et al (2017) NOTCH1 is a mechanosensor in adult arteries. Nat Commun 8:1620
Fang JS, Coon BG, Gillis N, Chen Z, Qiu J, Chittenden TW, Burt JM, Schwartz MA, Hirschi KK (2017) Shear-induced Notch-Cx37-p27 axis arrests endothelial cell cycle to enable arterial specification. Nat Commun 8:2149
Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D et al (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161:1163–1177
Lohela M, Bry M, Tammela T, Alitalo K (2009) VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol 21:154–165
Phng LK, Gerhardt H (2009) Angiogenesis: a team effort coordinated by notch. Dev Cell 16:196–208
Eilken HM, Adams RH (2010) Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22:617–625
Potente M, Carmeliet P (2017) The link between angiogenesis and endothelial metabolism. Annu Rev Physiol 79:43–66
Pitulescu ME, Schmidt I, Benedito R, Adams RH (2010) Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc 5:1518–1534
Benedito R, Hellström M (2013) Notch as a hub for signaling in angiogenesis. Exp Cell Res 319(9):1281–1288
Tammela T, Zarkada G, Nurmi H, Jakobsson L, Heinolainen K, Tvorogov D, Zheng W, Franco CA, Murtomaki A, Aranda E et al (2011) VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol 13:1202–1213
Siekmann AF, Lawson ND (2007) Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445:781–784
Shin M, Beane TJ, Quillien A, Male I, Zhu LJ, Lawson ND (2016) Vegfa signals through ERK to promote angiogenesis, but not artery differentiation. Development 143:3796–3805
del Toro R, Prahst C, Mathivet T, Siegfried G, Kaminker JS, Larrivee B, Breant C, Duarte A, Takakura N, Fukamizu A et al (2010) Identification and functional analysis. Blood 116:4025–4033
Ehling M, Adams S, Benedito R, Adams RH (2013) Notch controls retinal blood vessel maturation and quiescence. Development 140:3051–3061
Lim R, Sugino T, Nolte H, Andrade J, Zimmermann B, Shi C, Doddaballapur A, Ong YT, Wilhelm K, Fasse J, Ernst A, Kaulich M, Husnjak K, Boettger T, Guenther S, Braun T, Krüger M, Benedito R, Dikic I, Potente M (2019) Deubiquitinase USP10 regulates Notch signaling in the endothelium. Science 364(6436):188–193
Arima S, Nishiyama K, Ko T, Arima Y, Hakozaki Y, Sugihara K, Koseki H, Uchijima Y, Kurihara Y, Kurihara H (2011) Angiogenic morphogenesis driven by dynamic and heterogeneous collective endothelial cell movement. Development 138:4763–4776
Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A et al (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12:943–953
Kageyama R, Niwa Y, Shimojo H, Kobayashi T, Ohtsuka T (2010) Ultradian oscillations in Notch signaling regulate dynamic biological events. Notch signaling. Elsevier, Amsterdam, pp 311–331
Aspalter IM, Gordon E, Dubrac A, Ragab A, Narloch J, Vizan P, Geudens I, Collins RT, Franco CA, Abrahams CL et al (2015) Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting downstream of Notch. Nat Commun 6:7264
Mouillesseaux KP, Wiley DS, Saunders LM, Wylie LA, Kushner EJ, Chong DC, Citrin KM, Barber AT, Park Y, Kim JD et al (2016) Notch regulates BMP responsiveness and lateral branching in vessel networks via SMAD6. Nat Commun 7:13247
Moya IM, Umans L, Maas E, Pereira PN, Beets K, Francis A, Sents W, Robertson EJ, Mummery CL, Huylebroeck D, Zwijsen A (2012) Stalk cell phenotype depends on integration of Notch and Smad1/5 signaling cascades. Dev cell 22(3):501–514
Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, Stanchi F, Jones M, Aspalter IM, Cagna G et al (2014) The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol 16:309–321
Franco CA, Jones ML, Bernabeu MO, Geudens I, Mathivet T, Rosa A, Lopes FM, Lima AP, Ragab A, Collins RT et al (2015) Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol 13:e1002125
Panin VM, Papayannopoulos V, Wilson R, Irvine KD (1997) Fringe modulates Notch-ligand interactions. Nature 387(6636):908–912
Yang LT, Nichols JT, Yao C, Manilay JO, Robey EA, Weinmaster G (2005) Fringe glycosyltransferases differentially modulate Notch1 proteolysis induced by Delta1 and Jagged1. Mol Biol Cell 16:927–942
Williams CK, Li JL, Murga M, Harris AL, Tosato G (2006) Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 107(3):931–939
Harrington LS, Sainson RC, Williams CK, Taylor JM, Shi W, Li JL, Harris AL (2008) Regulation of multiple angiogenic pathways by Dll4 and Notch in human umbilical vein endothelial cells. Microvasc Res 75(2):144–154
Hogan BM, Herpers R, Witte M, Helotera H, Alitalo K, Duckers HJ, Schulte-Merker S (2009) Vegfc/Flt4 signalling is suppressed by Dll4 in developing zebrafish intersegmental arteries. Development 136:4001–4009
Sanganalmath SK, Bolli R (2013) Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ Res 113:810–834
Taimeh Z, Loughran J, Birks EJ, Bolli R (2013) Vascular endothelial growth factor in heart failure. Nat Rev Cardiol 10:519–530
Potente M, Mäkinen T (2017) Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol 18(8):477–494
Armulik A, Abramsson A, Betsholtz C (2005) Endothelial/pericyte interactions. Circ Res 97:512–523
Gaengel K, Genove G, Armulik A, Betsholtz C (2009) Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol 29:630–638
Cuervo H, Nielsen CM, Simonetto DA, Ferrell L, Shah VH, Wang RA (2016) Endothelial notch signaling is essential to prevent hepatic vascular malformations in mice. Hepatology 64:1302–1316
Dill MT, Rothweiler S, Djonov V, Hlushchuk R, Tornillo L, Terracciano L, Meili-Butz S, Radtke F, Heim MH, Semela D (2012) Disruption of Notch1 induces vascular remodeling, intussusceptive angiogenesis, and angiosarcomas in livers of mice. Gastroenterology 142:967–977
Dou GR, Wang YC, Hu XB, Hou LH, Wang CM, Xu JF, Wang YS, Liang YM, Yao LB, Yang AG et al (2008) RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J 22:1606–1617
Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, Niessen K, Plowman GD (2010) Chronic DLL4 blockade induces vascular neoplasms. Nature 463:E6-7
Couch JA, Zhang G, Beyer JC, de Zafra CL, Gupta P, Kamath AV, Lewin-Koh N, Tarrant J, Allamneni KP, Cain G et al (2016) Balancing efficacy and safety of an anti-DLL4 antibody through pharmacokinetic modulation. Clin Cancer Res 22:1469–1479
Noseda M, Chang L, McLean G, Grim JE, Clurman BE, Smith LL, Karsan A (2004) Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: role of p21Cip1 repression. Mol Cell Biol 24:8813–8822
Rostama B, Turner JE, Seavey GT, Norton CR, Gridley T, Vary CP, Liaw L (2015) DLL4/Notch1 and BMP9 interdependent signaling induces human endothelial cell quiescence via P27KIP1 and thrombospondin-1. Arterioscler Thromb Vasc Biol 35:2626–2637
Serra H, Chivite I, Angulo-Urarte A, Soler A, Sutherland JD, Arruabarrena-Aristorena A, Ragab A, Lim R, Malumbres M, Fruttiger M et al (2015) PTEN mediates Notch-dependent stalk cell arrest in angiogenesis. Nat Commun 6:7935
Sundlisaeter E, Edelmann RJ, Hol J, Sponheim J, Kuchler AM, Weiss M, Udalova IA, Midwood KS, Kasprzycka M, Haraldsen G (2012) The alarmin IL-33 is a notch target in quiescent endothelial cells. Am J Pathol 181:1099–1111
Polacheck WJ, Kutys ML, Yang J, Eyckmans J, Wu Y, Vasavada H, Hirschi KK, Chen CS (2017) A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 552(7684):258–262
Limbourg A, Ploom M, Elligsen D, Sörensen I, Ziegelhoeffer T, Gossler A, Drexler H, Limbourg FP (2007) Notch ligand Delta-like 1 is essential for postnatal arteriogenesis. Circ Res 100(3):363–371
Takeshita K, Satoh M, Ii M, Silver M, Limbourg FP, Mukai Y, Rikitake Y, Radtke F, Gridley T, Losordo DW, Liao JK (2007) Critical role of endothelial Notch1 signaling in postnatal angiogenesis. Circ Res 100(1):70–78
Cristofaro B, Shi Y, Faria M, Suchting S, Leroyer AS, Trindade A, Duarte A, Zovein AC, Iruela-Arispe ML, Nih LR et al (2013) Dll4-Notch signaling determines the formation of native arterial collateral networks and arterial function in mouse ischemia models. Development 140:1720–1729
Nus M, Martinez-Poveda B, MacGrogan D, Chevre R, D’Amato G, Sbroggio M, Rodriguez C, Martinez-Gonzalez J, Andres V, Hidalgo A et al (2016) Endothelial Jag1-RBPJ signalling promotes inflammatory leucocyte recruitment and atherosclerosis. Cardiovasc Res 112:568–580
Rafii S, Butler JM, Ding BS (2016) Angiocrine functions of organ-specific endothelial cells. Nature 529:316–325
Crivellato E, Nico B, Ribatti D (2007) Contribution of endothelial cells to organogenesis: a modern reappraisal of an old Aristotelian concept. J Anat 211(4):415–427
Matsumoto K, Yoshitomi H, Rossant J, Zaret KS (2001) Liver organogenesis promoted by endothelial cells prior to vascular function. Science 294(5542):559–563
Guo P, Poulos MG, Palikuqi B, Badwe CR, Lis R, Kunar B, Ding BS, Rabbany SY, Shido K, Butler JM, Rafii S (2017) Endothelial Jagged-2 sustains hematopoietic stem and progenitor reconstitution after myelosuppression. J Clin Investig 127(12):4242–4256
Poulos MG, Guo P, Kofler NM, Pinho S, Gutkin MC, Tikhonova A, Aifantis I, Frenette PS, Kitajewski J, Rafii S, Butler JM (2013) Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep 4(5):1022–1034
Gamrekelashvili J, Giagnorio R, Jussofie J, Soehnlein O, Duchene J, Briseño CG, Ramasamy SK, Krishnasamy K, Limbourg A, Kapanadze T, Ishifune C, Hinkel R, Radtke F, Strobl LJ, Zimber-Strobl U, Napp LC, Bauersachs J, Haller H, Yasutomo K, Kupatt C, Limbourg FP (2016) Regulation of monocyte cell fate by blood vessels mediated by Notch signalling. Nat Commun 7:12597
Krishnasamy K, Limbourg A, Kapanadze T, Gamrekelashvili J, Beger C, Häger C, Lozanovski VJ, Falk CS, Napp LC, Bauersachs J, Mack M, Haller H, Weber C, Adams RH, Limbourg FP (2017) Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat Commun 8(1):952
Ramasamy SK, Kusumbe AP, Wang L, Adams RH (2014) Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 507(7492):376–380
Duan JL, Ruan B, Yan XC, Liang L, Song P, Yang ZY, Liu Y, Dou KF, Han H, Wang L (2018) Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 68:677–690
Hilscher MB, Sehrawat T, Arab JP, Zeng Z, Gao J, Liu M, Kostallari E, Gao Y, Simonetto DA, Yaqoob U, Cao S, Revzin A, Beyder A, Wang RA, Kamath PS, Kubes P, Shah VH (2019) Mechanical stretch increases expression of CXCL1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology 157(1):193-209.e9
Kivelä R, Hemanthakumar KA, Vaparanta K, Robciuc M, Izumiya Y, Kidoya H, Takakura N, Peng X, Sawyer DB, Elenius K, Walsh K, Alitalo K (2019) Endothelial cells regulate physiological cardiomyocyte growth via VEGFR2-mediated paracrine signaling. Circulation 139(22):2570–2584
Cao Z, Lis R, Ginsberg M, Chavez D, Shido K, Rabbany SY, Fong GH, Sakmar TP, Rafii S, Ding BS (2016) Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat Med 22(2):154–162
Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, Wallenborg K, Picelli S, Zagorska A, Pendem SV et al (2019) Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep 29:1832–2184
Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, Geldhof V, de Rooij L, Kalucka J, Sokol L et al (2020) An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell 37:21–36
Guo M, Du Y, Gokey JJ, Ray S, Bell SM, Adam M, Sudha P, Perl AK, Deshmukh H, Potter SS et al (2019) Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat Commun 10:37
Kalucka J, de Rooij L, Goveia J, Rohlenova K, Dumas SJ, Meta E, Conchinha NV, Taverna F, Teuwen LA, Veys K et al (2020) Single-cell transcriptome atlas of murine endothelial cells. Cell 180:764–779
Karaiskos N, Rahmatollahi M, Boltengagen A, Liu H, Hoehne M, Rinschen M, Schermer B, Benzing T, Rajewsky N, Kocks C et al (2018) A Single-cell transcriptome atlas of the mouse glomerulus. J Am Soc Nephrol 29:2060–2068
Khan S, Taverna F, Rohlenova K, Treps L, Geldhof V, de Rooij L, Sokol L, Pircher A, Conradi LC, Kalucka J et al (2019) EndoDB: a database of endothelial cell transcriptomics data. Nucleic Acids Res 47:D736–D744
Menon R, Otto EA, Hoover P, Eddy S, Mariani L, Godfrey B, Berthier CC, Eichinger F, Subramanian L, Harder J et al (2020) Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight. https://doi.org/10.1172/jci.insight.133267
Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA et al (2019) Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575:512–518
Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Dominguez A, Pinho S, Akhmetzyanova I, Gao J, Witkowski M et al (2019) The bone marrow microenvironment at single-cell resolution. Nature 569:222–228
Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L et al (2018) A molecular atlas of cell types and zonation in the brain vasculature. Nature 554:475–480
Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE et al (2017) Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542:352–356
Acknowledgments
Work in Rui Benedito laboratory was supported by the Ministerio de Ciencia y Innovación (SAF2017-89299-P) and European Research Council (ERC-2020-COG n° 101001814). Macarena Fernandez-Chacon was supported by a PhD fellowship from Fundación La Caixa (CX_E-2015-01), Irene Garcia-Gonzalez was supported by La Caixa Severo Ochoa (CX-SO-16-1) and Severin Mühleder was funded by the Austrian Science Fund (FWF) project J4358. The CNIC is currently supported by MCIN and the Pro CNIC Foundation.
Author information
Authors and Affiliations
Contributions
MF-C, SM, IG-G and RB performed literature search and gave critical input. MF-C designed the figures. MF-C and RB wrote most of the manuscript. All authors read and approved the manuscript.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fernández-Chacón, M., García-González, I., Mühleder, S. et al. Role of Notch in endothelial biology. Angiogenesis 24, 237–250 (2021). https://doi.org/10.1007/s10456-021-09793-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-021-09793-7