
Abstract
Semaphorin 4D (SEMA4D) is a member of a family of transmembrane and secreted proteins that have been shown to act through its receptor Plexin-B1 to regulate axon growth cone guidance, lymphocyte activation, and bone density. SEMA4D is also overexpressed by some malignancies and plays a role in tumor-induced angiogenesis similar to vascular endothelial growth factor (VEGF), a protein that has been targeted as part of some cancer therapies. In an attempt to examine the different effects on tumor growth and vascularity for these two pro-angiogenic factors, we previously noted that while inhibition of both VEGF and SEMA4D restricted tumor vascularity and size, vessels forming under conditions of VEGF blockade retained their association with pericytes while those arising in a background of SEMA4D/Plexin-B1 deficiency did not, an intriguing finding considering that alteration in pericyte association with endothelial cells is an emerging aspect of anti-angiogenic intervention in the treatment of cancer. Here we show through array analysis, immunoblots, migration and co-culture assays and VE-cadherin immunohistochemistry that SEMA4D production by head and neck carcinoma tumor cells induces expression of platelet-derived growth factor-B and angiopoietin-like protein 4 from endothelial cells in a Plexin-B1/Rho-dependent manner, thereby influencing proliferation and differentiation of pericytes and vascular permeability, whereas VEGF lacks these effects. These results partly explain the differences observed between SEMA4D and VEGF in pathological angiogenesis and suggest that targeting SEMA4D function along with VEGF could represent a novel anti-angiogenic therapeutic strategy for the treatment of solid tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plexins are transmembrane receptors that regulate cell motility and adhesion in a variety of tissue types. There are nine known plexins, Plexin-A1 to A4, -B1 to B3, -C1 and -D1, grouped according to domain structure, which bind their primary ligands, the semaphorins (SEMAs) [1]. The SEMAs are grouped into eight classes: Class I, IV, V, and VI cross the membrane, class VII is GPI-linked directly to the membrane, and classes II and III are secreted (the final class is a viral SEMA). Some class III SEMAs require neuropilins (NPs) as co-receptors, which are also known to participate in vascular endothelial growth factor (VEGF) signaling [2]. Promiscuity in binding exists between different SEMA classes and the various plexins, though binding partners and their effects on cell physiology are likely modulated in a cell-specific manner. SEMAs and plexins are known to influence a wide range of processes in both health and disease including the development and regeneration of the nervous and cardiovascular systems, bone development and density, and activation of lymphocytes [3]. Our lab and others have shown that Semaphorin 4D (SEMA4D) is pro-angiogenic when acting through its receptor Plexin-B1 on endothelial cells [4, 5] and may be produced by malignancies for the purposes of promoting blood vessel growth into the tumor in a manner analogous to VEGF [6]. Both SEMA4D and VEGF are potent pro-angiogenic, pro-survival factors upregulated in malignancies under conditions of hypoxia [7].
The role of VEGF and its receptor VEGFR-2 in tumor-induced angiogenesis has been well characterized, but less is known about the effects of other factors expressed during cancer progression, such as SEMA4D. In an effort to compare the contributions of SEMA4D and VEGF to tumor growth and vascularity, we altered production of both proteins alone or in combination with RNA interference, blocking antibodies and through manipulation of the hypoxia response and noted a reduction in tumor size and vascularity [8, 9]. However, vessels from tumors composed of cells with inhibited VEGF retained a close association with pericytes, similar to what others have reported [10], while tumors composed of cells in a background of SEMA4D blockade lacked this feature [8]. These findings led us to investigate the inherent difference in how these factors influence recruitment of pericytes and control vascular maturity and permeability.
Here, through the use of arrays, immunoblot analysis, in vitro and in vivo measurements of angiogenesis and VE-cadherin immunohistochemistry, we demonstrate that soluble SEMA4D and SEMA4D derived from head and neck squamous cell carcinoma (HNSCC) cell lines drives endothelial production of platelet derived growth factor (PDGF)-B and angiopoietin-like 4 (ANGPTL4) in a Plexin-B1/RhoA-dependent manner, an effect we failed to observe with VEGF. PDGF-B is a crucial player in differentiation and chemotaxis of pericytes, which express its receptor PDGFR-β and respond by associating with endothelial cells in blood vessels [11]. The role of tumors in this process is not well described, even though failure of anti-VEGF/VEGFR-2 therapy may be linked to protection of newly formed tumor vessels by pericyte sheaths [12, 13]. Even less is known about ANGPTL4. First identified in adipose tissue where it was shown to inhibit lipoprotein lipase and raise plasma triglyceride levels [14, 15], recent studies have demonstrated that this protein is upregulated in tumors, including HNSCC, also under conditions of hypoxia [16–19]. ANGPTL4 induces vascular permeability by interfering with VE-cadherin function, thereby promoting angiogenesis, influencing tumor survival and enhancing metastasis [17, 20, 21].
A new concept in anti-angiogenic therapy is emerging involving combined targeting of endothelial cells and pericytes. This strategy might be able to prevent angiogenesis through inhibition of vessel stabilization, while at the same time suppressing metastatic potential [13]. The results presented here highlight mechanistic differences between SEMA4D and VEGF in tumor-induced angiogenesis and suggest that SEMA4D blockade could be an excellent form of treatment for some malignancies concurrent with anti-VEGF therapy, or where anti-VEGF therapy has failed to achieve a desired outcome.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVEC, ATCC, Manassas, VA) were cultured in Endothelial Cell Medium-2 (EGM-2, Lonza, Basel, Switzerland). 293T (ATCC) cells and HNSCC cell lines [22] were cultured in DMEM (Sigma, St. Louis, MO) supplemented with 10 % fetal bovine serum (unless otherwise indicated) and 100 units/ml penicillin/streptomycin/amphotericin B (Sigma). The human pericyte line hPC-PL (PromoCell, Heidelberg, Germany) were grown in pericyte growth medium (PromoCell) and C3H/10T1/2 embryonic mesenchymal stem cells (a gift from Dr. Snigdha Banerjee [23]) were grown in DMEM supplemented with 10 % fetal bovine, 233.6 μg/ml glutamine, 25 mM glucose, and 100 units/ml penicillin/streptomycin/amphotericin, and treated as indicated.
Purification of soluble SEMA4D
Soluble SEMA4D (sSEMA4D) was produced and purified as described previously [4]. Briefly, the extracellular portion of SEMA4D was subjected to PCR and the resulting product cloned into the plasmid pSecTag2B (Invitrogen, Carlsbad, CA). This construct was transfected into 293T cells growing in serum free media. Media containing sSEMA4D was collected 65 h post-transfection and purified with TALON metal affinity resin (Clontech Laboratories, Palo Alto, CA) according to manufacturer’s instructions. Concentration and purity of the TALON eluates was determined by SDS PAGE analysis followed by silver staining (Amersham Life Science, Piscataway, NJ) and the Bio-Rad protein assay (Bio-Rad, Hercules, CA). In all cases, media collected from cells transfected with the empty pSecTag2B vector were used as control.
Angiogenesis arrays
Antibody-based angiogenesis arrays were purchased from RayBiotech (Norcross, GA), with experiments performed according to manufacturer’s instructions. Briefly, 1 × 106 HUVECs were grown in serum free media in a 100 mm tissue culture plate overnight and then treated with 100 ng/ml of VEGF or 800 ng/ml of sSEMA4D for 2 h, lysed in 1X RayBiotech Cell Lysis Buffer, and 50 μg of lysate applied to the protein array slides (sandwich ELISAs) containing antibodies for specific angiogenesis-associated proteins. Slides were processed at RayBiotech with fluorescent dye-conjugated streptavidin, scanned, and relative pixel intensity determined.
ELISA assay
Confluent HUVECs were serum starved for 4 h, then cultured in serum free medium with 800 ng/ml SEMA4D. Conditioned media were collected at the indicated time points and used to analyze PDGF-B and ANGPTL4 production by ELISA (Cytokine Core Facility, University of Maryland School of Medicine). Results are expressed as the average and standard deviation for three independent experiments.
Rho pull-down assay
HUVEC were washed in PBS and grown in serum free medium for 36 h. The cells were then washed in PBS and treated with 800 ng/ml sSEMA4D with or without 6 μg/ml C3 toxin (List Biological Laboratories, Campbell, CA) for the time points indicated. Cells were washed with PBS and lysed in lysis buffer [50 mM Tris–HCl, 150 mM NaCl, 1 % NP 40 supplemented with protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride, 1 μl/ml aprotinin and leupeptin, Sigma) and phosphatase inhibitors (2 mM NaF and 0.5 mM sodium orthovanadate, Sigma)]. Rho activity was assessed using purified glutathione S-transferase- rhotekin-RBD previously bound to glutathione-Sepharose 4B (Amersham Pharmacia, Sweden) to affinity precipitate GTP-bound RhoA. Western blot analysis of total and active Rho was performed using a monoclonal antibody against RhoA (Santa Cruz Biotechnology; 26C4). Immuno-complexes were visualized by enhanced chemiluminescence detection (Amersham Life Science) using goat anti-mouse coupled to horseradish peroxidase as a secondary antibody (Santa Cruz).
Immunoblots
Cells grown in serum free media, 10 % serum, or treated with sSEMA4D or VEGF, as indicated, were lysed in lysis buffer (see above) for 15 min at 4 °C. After centrifugation, protein concentrations were measured using the Bio-Rad protein assay (Bio-Rad). 100 μg of protein from each sample was subjected to SDS–polyacrylamide gel electrophoresis and transferred onto a PVDF membrane (Immobilon P, Millipore Corp., Billerica, MA). The membranes were then incubated with the following antibodies: anti-PDGF-B (LifeSpan Biosciences, Seattle, WA); anti-ANGPTL4 (Chemicon/Millipore); anti-GAPDH (Sigma); anti-RGS5 (Sigma); anti-αSMA (Abcam, Cambridge, MA ab5694); anti-αNG2 (Thermo Scientific, Waltham, MA) [24]; anti-Plexin-B1 (A8, Santa Cruz Biotech, Santa Cruz, CA); anti-SEMA4D (BD Transduction Labs, BD Biosciences, Palo Alto, CA). Proteins were detected using the ECL chemiluminescence system (Pierce, Rockford, IL). Where indicated, cells were treated with 6 μg/ml C3 toxin (List Biological Laboratories) or 4 μg/ml isotype-matched control IgG or anti-PDGF-B antibody.
Short hairpin (sh) RNA and lentivirus infections
The shRNA sequences for human SEMA4D and Plexin-B1 were obtained from Cold Spring Harbor Laboratory’s RNAi library (RNAi Central, http://cancan.cshl.edu/RNAi_central/RNAi.cgi?type=shRNA) [25, 26]. The sequences used as PCR templates for SEMA4D and Plexin-B1 shRNA have been previously reported [6, 27]. Oligos were synthesized (Invitrogen) and cloned into pWPI GW, a Gateway compatible CSCG based lentiviral destination vector as previously described [6, 28, 29]. Viral stocks were prepared and infections performed as previously reported [6].
RNA isolation and RT-PCR analysis
RNA was extracted from HUVEC cell lysates, controls or those treated with sSEMA4D or VEGF, with or without C3 toxin, and converted into cDNA using the AMV reverse-transcriptional system (Promega, Madison, WI) in the presence of random hexamers (Invitrogen). The cDNA was used for quantitative real-time PCR with specific gene primers as follows: PDGF-B forward: 5′- AACAACCGCAACGTGCAGTG -3′, reverse: 5′- CCGAATGGTCACCCGAGTTT -3′; ANGPTL4 forward: 5′- TGGGTCTGGAGA AGGTGCATA -3′, reverse: 5′- CTGGCCGTTGAGGTTGGAAT -3′; and 18S forward: 5′- TTGACGGAAGGGCACCACCAG -3′, reverse: 5′- GCACCACCACCCACGGAATCG -3′. An MYIQ real-time PCR detection system and SYBR green PCR mix (Bio-Rad) were used to carry out real-time PCR. The relative abundance of transcript was quantified using the comparative Ct method with 18S as an internal control. All data were analyzed from three independent experiments and statistical significance validated by Student’s t test.
Proliferation assays
Cell proliferation rate was determined using CCK-8 (Dojindo Molecular Technologies, Gaithersburg, MD), per manufacturer’s instructions. Cells were grown in 100 μl of culture medium and placed into 96-well plates at a concentration of 3 × 103 cells/well. Cells then were incubated with 5 μl of CCK-8 for 1 h at 37 °C. Absorbance at 450 nm was measured using a plate reader.
In vitro migration assays
Serum free media conditioned by HUVECs or head and neck (HN)12 and HN13 cells, control infected or infected with lentivirus expressing Plexin-B1 shRNA and treated with 800 ng/ml sSEMA4D with or without 4 μg/ml IgG or anti-PDGF-B antibody, where indicated, were used as the chemoattractants for C3H/10T1/2 or hPC-PL cells migrating through a polyvinylpyrrolidone membrane (8 μ pore size, Osmonics; GE Water Technologies, Trevose, PA) in a Boyden chamber assay, as previously described [4]. 0.1 % BSA was used as the negative control. Cell migration was expressed as pixel intensity of stained, scanned membranes. Each experiment was performed in triplicate and average and standard deviation calculated.
Pericyte and endothelial cell co-culture assays
Co-culture of hPC-PL with HUVEC was performed on Cultrex reconstituted basement membrane extract (Trevigen, Gaithersburg, MD), as previously described [30]. Briefly, cells were grown in DMEM with or without 4 μg/ml IgG or anti-PDGF-B antibody, 6 μg/ml C3 toxin (List Biological Laboratories), or where endothelial cell expression of Plexin-B1 was silenced with shRNA (see above), for 48 h in the presence of 800 ng/ml sSEMA4D or media conditioned by HN12 or HN13 cells. Cells were fixed with 4 % paraformaldehyde and stained using anti-CD31 antibody (Millipore; 1:200 dilution) and PDGFR-β (Abcam, 1:200 dilution). Secondary antibodies were FITC-goat anti-rat (Santa Cruz) and Texas red-goat anti-rabbit (Abcam). Slides were examined with a Nikon Eclipse E800 microscope system.
Tumor cell injections and animal studies
1 × 106 HN12 or HN13 cells, controls or infected ex vivo with lentiviruses coding for SEMA4D shRNA or Plexin-B1 shRNA, were resuspended in 100 μl of serum-free DMEM with an equal volume of liquid Cultrex basement membrane extract (Trevigen) and injected subcutaneously into the flanks of immunocompromised nude mice. Animals were euthanized and tumors removed for sectioning and processing for immunofluorescence (see below). All animal studies were approved by the University of Maryland Office of Animal Welfare, Institutional Animal Care and Use Committee, in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Endothelial cell and pericyte immunofluorescence
Tumor tissues were processed for co-immunofluorescence as described [31]. Briefly, OCT-embedded 8 μm thick frozen tissue sections were cut onto silanated glass slides, air-dried, and stored at −80 °C. Cryosections were thawed, hydrated, fixed, blocked in 10 % FBS, and incubated overnight at 4 °C with primary antibodies diluted in a 2 % BSA/0.1 % Tween 20 solution in PBS. The following primary antibodies were used: anti-CD31 (anti-PECAM, BD Pharmingen; 1:100 dilution); anti-PDGFR-β (Abcam, 1:200 dilution). After washing with PBS, the slides were incubated with FITC-conjugated anti-rat (Sigma) and Texas red-conjugated anti-rabbit (Calbiochem, EMD Biosciences, San Diego, CA) secondary antibodies for 1 h at room temperature. Finally, the slides were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) and examined with a Nikon Eclipse E800 microscope system. Blood vessel and pericyte density was determined by counting the number of stained vessels in 10 fields at a magnification of 100× and calculating the average and standard deviation.
VE-cadherin internalization and immunofluorescence
VE-cadherin internalization was tracked using the anti-VE-cadherin BV6 clone antibody uptake assay. Sub-confluent HUVEC cultures, controls or with silenced ANGPTL4, were first incubated with BV6 antibody (1:200 dilution; Enzo Life Sciences) at 4 °C for 1 h to label surface VE-cadherin. Samples were then washed with ice-cold PBS and treated with serum free DMEM containing 100 ng/ml VEGF or 800 ng/ml sSEMA4D for 8 h at 37 °C. After extensive PBS washes, cells were either fixed in 4 % paraformaldehyde for 15 min or subjected to acid wash in 0.1 M Glycine, pH 2.7, for 15 min at room temperature prior to fixation. Cells were permeabilized with 0.5 % PBS-Triton X-100 for 5 min and blocked in 3 % PBS-BSA for 45 min, followed by 1 h incubation with Alexa Fluro 555 goat anti-mouse IgG2a (Invitrogen), 1:400 dilution in 3 % PBS-BSA. Samples were then stained with DAPI (1:1,000 dilution in PBS; Invitrogen) for 10 min, mounted in Mounting Medium (Vector Laboratories), and analyzed by a Nikon Eclipse 800 microscope.
Statistical analysis
Student’s paired t tests were performed on means, and p values calculated: * p ≤ 0.05; ** p ≤ 0.01.
Results
Treatment of endothelial cells with SEMA4D, but not VEGF, elicits production of PDGF-B and ANGPTL4 in a Rho-dependent manner
Having noticed differences between SEMA4D and VEGF on malignant cell proliferation, tumor growth, and the quality of tumor vasculature, including association of endothelial cells with pericytes and hence vessel stability and maturity [8], we wanted to examine the effects of these two proteins on endothelial cell production of pro-angiogenic factors. HUVECs treated for 2 h with soluble SEMA4D (sSEMA4D) examined by an ELISA angiogenesis array revealed induction of PDGF-B and ANGPTL4, relative to controls (Fig. 1a, upper right panel, upregulated proteins indicated in red), whereas VEGF failed to show a similar response (Fig. 1a, lower right panel, unchanged proteins in yellow). IL-8 production also increased in the presence of sSEMA4D, which was expected since we have observed this effect before [27] and therefore served as an internal control (Fig. 1a). Interestingly, VEGF treatment of HUVECs caused an increase in VEGF production from these cells in the array. We suspect that this might be residual VEGF bound to cells following treatment and not true VEGF production, but it remains a possibility. The results of the array are quantified by fluorescence intensity, relative to negative controls, in Fig. 1b. The array studies were done using cell lysates, so we wanted to confirm production of soluble protein with an ELISA on media conditioned by HUVEC incubated with sSEMA4D and VEGF. These results confirm production of PDGF-B and ANGPTL4 (Fig. 2c, left and right panels respectively) compared to controls, starting at about 2 h post-treatment.

Soluble SEMA4D, but not VEGF, elicits production of PDGF-B and ANGPTL4 from HUVECs in a Rho-dependent manner. a Angiogenesis array performed on control HUVEC (left panel) and HUVEC treated with soluble SEMA4D (sSEMA4D, upper right panel) or VEGF (lower right panel) for 2 h. Proteins of interest are highlighted in blue in untreated cells. sSEMA4D resulted in upregulation of PDGF-B, ANGPTL4 and IL-8 (red), while treatment with VEGF did not change levels of these proteins (yellow). Elevated levels of VEGF were detected in VEGF treated HUVECs, possibly as a result of residual VEGF binding. Positive and negative controls are in white. b Quantification of array fluorescence for PDGF-B and ANGPTL4 relative to negative controls. c ELISA for production of PDGF-B (left panel, in pg/ml) and ANGPTL4 (right panel, ng/ml) in HUVEC treated with sSEMA4D for the indicated times. Results are expressed as averages and error bars represent the standard deviation for three independent experiments. d Control HUVECs (C) or HUVECs treated with sSEMA4D alone (C) or with the RhoA signaling pathway inhibitor C3 toxin (C3) for the indicated times were subjected to a Rho pulldown assay. Active, GTP-bound RhoA (top panel) was lost in cells incubated with C3 toxin. Total Rho was used as a loading control (lower panel). e Immunoblot of HUVECs, controls (C) or treated for 24 and 48 h with VEGF or sSEMA4D alone (C) or with C3 toxin (C3) confirms upregulation of PDGF-B (upper panel) and ANGPTL4 (middle panel) for sSEMA4D only, occurring in a RhoA-dependent manner. GAPDH was used as a loading control (lower panel). f RT-PCR confirmation of array and immunoblot results reveals that sSEMA4D-induced increases in PDGF-B and ANGPTL4 occur at the level of mRNA in a Rho-dependent manner (* p ≤ 0.05; ** p ≤ 0.01 for all graphs)

SEMA4D-induced production of PDGF-B from endothelial cells causes differentiation, proliferation and migration of pericytes and their association with HUVECs. a C3H/10T1/2 were grown in media conditioned by HUVECs with and without serum, treated with sSEMA4D with or without anti-PDGF-B blocking antibody, and differentiation determined by immunoblot for RGS5, NG2, and αSMA. GAPDH was used as a loading control. b Immunoblot for RGS5, NG2, and αSMA in C3H/10T1/2 cells grown in media conditioned by sSEMA4D treated HUVECs, either controls (C) or HUVEC with Plexin-B1 silenced by shRNA (PB1shRNA). c Proliferation of C3H/10T1/2 cells determined by optical density of stained cells at 450 nm (Y-axis) at 0, 2 and 4 days in media conditioned by HUVECs with and without serum treated with sSEMA4D, with silenced Plexin-B1 (Plexin-B1 shRNA), control IgG or anti-PDGF-B blocking antibody (α-PDGF-B). d Migration of C3H/10T1/2 cells towards 0.1 % BSA (negative control) or media conditioned by untreated HUVECs (untreated) or HUVECs treated with sSEMA4D (sS4D) with or without silenced Plexin-B1 (PB1 shRNA), control IgG or anti-PDGF-B blocking antibody (α-PDGF-B). e Quantification of stained migration assay membrane, determined by pixel intensity (Y-axis). Error bars represent the standard deviation from three migrations per condition. f HUVECs (green) were co-cultured with hPC-PL cells (pericytes, red) in the presence of sSEMA4D (left three columns) or in media conditioned by HN12 cells (middle three columns) or HN13 cells (right three columns), either in control conditions (top row), or in the presence of anti-PDGF-B antibody (second row), where Plexin-B1 was silenced in HUVECs with shRNA (Plexin-B1 shRNA, third row) or incubated with C3 toxin (fourth row). Co-association of these cells is shown (merge, yellow). g Quantification of co-association assay (% counted HUVECs in 10 hpf associated with pericytes, Y-axis; * p ≤ 0.05; ** p ≤ 0.01 for all graphs). h Immunoblot for Plexin-B1 (upper panel) in 293T transfected with a Plexin-B1 construct, hPC-PL cells and HUVECs, to rule out direct effects of SEMA4D on pericytes. GAPDH was used a loading control for all blots (lower panels)
We and others have shown that when activated by SEMA4D, Plexin-B1 signals through Rho to elicit many of its effects [4, 32, 33]. To decipher this mechanism in endothelium, we treated HUVECs with sSEMA4D, with and without the Clostridium botulinum toxin C3, which is known to inhibit activation of Rho signaling pathways, and performed a pulldown assay for GTP-bound, active Rho. We observe that sSEMA4D specifically activates Rho signaling in HUVECs, an effect lost in cells incubated with C3 (Fig. 1d). To confirm the results of the ELISAs, and to demonstrate that upregulation of PDGF-B and ANGPTL4 require intact Rho signaling, we performed an immunoblot for PDGF-B and ANGPTL4 in HUVECs treated with VEGF and sSEMA4D, with and without C3 toxin. Similar to the array, cells incubated with VEGF failed to exhibit upregulation of these proteins while sSEMA4D did (Fig. 1e). However, co-treatment with C3 suppressed these effects at 24 and 48 h (Fig. 1e). These results were further confirmed by RT-PCR, demonstrating that this is a transcriptional effect (Fig. 1f). Taken together, these findings show that SEMA4D induces Plexin-B1/RhoA-dependent production of PDGF-B and ANGPTL4 from HUVECs while VEGF does not, suggesting a possible mechanism for differences of these two pro-angiogenic proteins observed in endothelial cells [8, 9].
SEMA4D-induced production of PDGF-B by HUVECs promotes differentiation of mesenchymal stem cells into pericytes and pericyte proliferation, chemotaxis, and association with HUVECs in a capillary network.
PDGF-B is a crucial factor in the recruitment of pericytes to newly formed vessels [11]. Both endothelial and non-endothelial cells may produce PDGF-B in physiological and pathological angiogenesis to induce differentiation of mesenchymal stem cells into pericytes and their association with blood vessels [34]. Therefore, to test the significance of SEMA4D-induced production of PDGF-B by endothelial cells, we used the mouse embryonic mesenchymal stem cell line C3H/10T1/2, isolated from C3 H mouse embryo pluripotent stem cells that have been shown to have the ability to differentiate into a variety of mesodermal lineages [23] and incubated them in media conditioned by HUVEC with and without serum, treated with sSEMA4D with and without anti-PDGF-B blocking antibody. Using expression of RGS5, αSMA [35] and NG2 [24] as indicators of pericyte differentiation, we observed a slight increase in expression of these proteins in serum alone and a more robust response in cells grow in media conditioned by sSEMA4D treated HUVECs (Fig. 2a). This effect was reduced in cells incubated with anti-PDGF-B blocking antibody (Fig. 2a). Differentiation was dependent upon Plexin-B1 function in HUVECs, as media conditioned by HUVEC where this receptor was knocked down with an shRNA-expressing lentivirus could not elicit RGS5, αSMA and NG2 upregulation in C3H/10T1/2 (Fig. 2b). To examine effects on proliferation, C3H/10T1/2 were grown in serum free media conditioned by control HUVECs, or HUVECs treated with sSEMA4D but with silenced Plexin-B1 or incubated with IgG or anti-PDGF-B blocking antibody. Proliferation was noted at 2 and 4 days only in populations where sSEMA4D could signal through Plexin-B1 in HUVEC and where PDGF-B in the medium was not blocked with antibody (Fig. 2c), indicating that SEMA4D/Plexin-B1-mediated production of PDGF-B by HUVEC was causing proliferation. To determine PDGF-B effects on migration, C3H/10T1/2 cells were used in a migration assay towards media containing 0.1 % BSA (negative control) or media conditioned by HUVEC, untreated or treated with sSEMA4D but control infected or infected with Plexin-B1 shRNA-expressing lentiviruses or where cells were incubated with IgG or anti-PDGF-B antibody, similar to the conditions in Fig. 2c. Migration was only observed towards media conditioned by HUVEC treated with sSEMA4D that had functional Plexin-B1 or where PDGF-B was not neutralized by antibody, when compared to controls (Fig. 2d). The results of the migration assay are quantified in Fig. 2e.
We next co-cultured HUVEC, controls or those with silenced Plexin-B1, with cells of the human pericyte line hPC-PL in the presence or absence of anti-PDGF-B blocking antibody, either in sSEMA4D or in media conditioned by HN12 or HN13 cells, looking for their co-association by immunofluorescence [30]. HUVECs (Fig. 2f, in green) and pericytes (red) exhibited association (merge, yellow) under conditions where Plexin-B1 was expressed in HUVECs and PDGF-B was not bound by blocking antibody, in both sSEMA4D (left three columns) or media conditioned by HN12 (middle three columns) or HN13 cells (right three columns), which are known to shed SEMA4D from the cell surface through proteolysis [36, 37]. Quantification of co-association of pericytes with HUVECs is shown in the graph in Fig. 2g. To determine if any of these observations could be the result of direct effects of SEMA4D on pericytes, we looked for the presence of Plexin-B1 on hPC-PL cells. Figure 2h demonstrates a lack of detectable Plexin-B1 in hPC-PL when compared to 293T cells transfected with a Plexin-B1 construct or HUVECs, which acted as positive controls. Taken together, these results demonstrate that PDGF-B produced by HUVEC in response to SEMA4D induces pericyte differentiation of stem cells, their proliferation and migration, and their association with HUVECs, suggesting a crucial role for SEMA4D in crosstalk between endothelial cells and pericytes for vascular development and maturity.
Effects of tumor cell SEMA4D/Plexin-B1 on vasculature
Some transformed cells express both SEMA4D and Plexin-B1 [38], so the possibility exists that malignant cells might be a source of PDGF-B and ANGPTL4 as a result of an autocrine or paracrine signaling mechanism that could influence endothelial-pericyte communication in pathological or tumor-induced angiogenesis. To test this and any effects such products might have on tumor vasculature, we first looked for production of these proteins in an immunoblot in HN12 cells with and without silenced SEMA4D and Plexin-B1. We confirmed knockdown of these proteins through infection with lentiviruses coding for the appropriate shRNAs in an immunoblot (Fig. 3a). Control HN12 cells express PDGF-B at baseline, the levels of which were reduced slightly in cells with either silenced SEMA4D or Plexin-B1 (Fig. 3b). Interestingly, ANGPTL4 was not expressed by HN12 under any conditions (Fig. 3b). To determine the ability to attract pericytes, we used media conditioned by control HN12 or HN13 tumor cells, cells with silenced Plexin-B1, or cells incubated with IgG or anti-PDGF-B blocking antibody as the chemoattractants for the pericyte line hPC-PL in Boyden chamber migration assays. Media conditioned by control infected and IgG treated HN cells induced hPC-PL migration compared to 0.1 % BSA (negative control, Fig. 3c). Cells with silenced Plexin-B1, which would disrupt SEMA4D autocrine or paracrine signaling and reduce PDGF-B production, exhibited no appreciable reduction in migration (Fig. 3c, PB1 shRNA), while media from HN12 and 13 incubated with anti-PDGF-B antibody (α-PDGFB) showed a slight inhibition in migration. Results of the migration assay are quantified in Fig. 3d. These findings suggest that autocrine or paracrine SEMA4D/Plexin-B1 signaling contributes only slightly to production of PDGF-B in tumor cells and that there are likely other mechanisms for its production and other chemoattractant proteins made by cancer cells that influence the migration of pericytes.

Effects of tumor cell SEMA4D on vasculature. a Lentiviruses expressing SEMA4D shRNA (S4D shRNA) or Plexin-B1 shRNA (PB1 shRNA) were used to silence expression of these proteins in HN12 cells, with knockdown confirmed by immunoblot (left and right panels, respectively). b Immunoblot for PDGF-B (upper panel) and ANGPTL4 (middle panel) in HN12 cells, control infected or infected with lentivirus expressing SEMA4D shRNA or Plexin-B1 shRNA from (a). c Migration assay to determine chemotaxis of hPC-PL cells toward media containing 10 % serum (positive control), 0.1 % BSA (negative control) or media conditioned by HN12 cells (top panel) or HN13 cells (bottom panel), control infected (control sh), those infected with shRNA-expressing lentivirus to silence Plexin-B1 (PB1 shRNA) or when treated with IgG or anti-PDGF-B blocking antibody (α-PDGF-B). d Quantification of stained migration assay membranes, determined by pixel intensity (Y-axis). Error bars represent the standard deviation from three migrations per condition. e HUVEC treated with media conditioned by HN12 cells were control infected (C) or infected with Plexin-B1-expressing lentivirus (left panel) or treated with carrier (control, C) or C3 toxin (C3) for 24 and 48 h and blotted or expression of PDGF-B (upper panel) and ANGPTL4 (middle panel). GAPDH was used as a loading control for all blots (bottom panels). (f) HN12 (left three columns) and HN13 (right three columns), control infected (C, top row) or infected with lentivirus coding for shRNAs to silence SEMA4D (S4D shRNA, middle row) or Plexin-B1 (PB1 shRNA, bottom row), were grafted into the flanks of nude mice and the resulting tumors analyzed by CD31 (green, left columns) and PDGFR-β (red, middle columns) expression for endothelial cells and pericytes, respectively. Association of endothelial cells with pericytes is shown in the third column (merge, right column). g Quantification of blood vessels and pericytes from the tumor xenografts shown in (f) demonstrates loss of endothelial cells and pericytes from tumors with silenced SEMA4D, but little effect on endothelial cell content and a slight inhibition of pericytes in tumors with silenced Plexin-B1 (* p ≤ 0.05; ** p ≤ 0.01 for all graphs)
Despite these results, we do know that HN12 cells and other tumor cells lines and tissues express SEMA4D [6], and we previously saw that sSEMA4D robustly increased expression of PDGF-B and ANGPTL4 in HUVEC well above baseline. We therefore wanted to examine the effects of tumor-derived sSEMA4D on production of these proteins in endothelial cells. We treated control HUVECs, those with silenced Plexin-B1, and HUVECs incubated with C3, with media conditioned by HN12 cells and looked for production of PDGF-B and ANGPTL4 in an immunoblot. We observed high levels of these proteins in controls, but not in HUVEC with silenced Plexin-B1, where SEMA4D would have no effect, or in cells growing in C3, which inactivates Rho signaling pathways (Fig. 3e). To determine the in vitro significance to these findings, we performed tumor xenografts in mice using control HN12 and 13 cells, those with silenced SEMA4D to disrupt Plexin-B1 signaling in the tumor and vessels of the stroma, and cells with silenced Plexin-B1, which would still elicit SEMA4D-mediated effects on endothelial cells (and then indirectly on pericytes) but fail to activate autocrine or paracrine signaling in the tumor, and examined tumor vascularity and pericyte content of the tumor stroma by immunofluorescence. We observed that silencing SEMA4D in HN cells resulted in reduced tumor vascularity and pericytes, as we have noted previously [8] (Fig. 3f). Silencing Plexin-B1 resulted in no appreciable effect on tumor endothelial cells and only a slight reduction in pericyte numbers (Fig. 3f). These results are quantified in the bar graph in Fig. 3g. Taken together these results show that SEMA4D/Plexin-B1 autocrine or paracrine signaling plays a small role in PDGF-B production by tumors and subsequent influence over vasculature, while tumor-derived SEMA4D very strongly induces production of PDGF-B and ANGPTL-4 in endothelial cells, influencing association of endothelial cells with pericytes.
SEMA4D/Plexin-B1-mediated induction of ANGPTL4 in HUVEC causes internalization of VE-cadherin
Originally identified in adipose tissues and liver and believed to be involved in lipid metabolism, ANGPTL4 is now known to be a multifaceted protein important in homoeostasis, wound repair, tumorigenesis, angiogenesis, and redox regulation [39]. Reports have suggested that ANGPTL4 is pro-angiogenic and pro-metastatic, enhancing both of these processes through disruption of VE-cadherin function in endothelial cells and weakening of cell–cell junctions [18, 20, 21]. To determine the biological significance of increased ANGPTL4 production by HUVECs in response to SEMA4D, we incubated control HUVECs or HUVECs with silenced ANGPTL4 (Fig. 4a) with VEGF and sSEMA4D and looked for effects on VE-cadherin expression. Control HUVECs demonstrated cell surface VE-cadherin, evidenced by loss of signal in immunofluorescence analysis of cells following acid washing, which removes cell surface proteins (Fig. 4b, first panel). As expected, VEGF, which is known to induce vascular leakiness through endothelial cell retraction, retained VE-cadherin immunofluorescence as it was internalized and protected from acid wash (second panel). VEGF-mediated internalization likely does not involve ANGPTL4, as its silencing had no effect (third panel). While sSEMA4D treatment resulted in the same response as VEGF, with VE-cadherin internalization and loss of cell–cell contacts (fourth panel), when ANGPTL4 was silenced, sSEMA4D failed to cause internalization of VE-cadherin, thereby preventing vascular permeability (last panel). These results indicate that ANGPTL4 is required for SEMA4D-mediated internalization of VE-cadherin and vascular permeability.

SEMA4D induction of ANGPTL4 in HUVEC causes internalization of VE-cadherin. a Immunoblot for HUVECs transfected with scrambled oligos (C) or ANGPTL4 siRNA demonstrates successful suppression of ANGPTL4 protein (upper panel). GAPDH is used as the loading control (lower panel). b HUVEC monolayer, untreated, or treated with VEGF or sSEMA4D with or without ANGPTL4 siRNA, stained for VE-cadherin (green) and acid washed to remove cell surface/membrane protein. HUVEC exhibit internalization of cadherin in VEGF or sSEMA4D compared to untreated controls, an effect lost in sSEMA4D treated cells with silenced ANGPTL4 (nuclei stained with DAPI, blue)
Discussion
A great deal of research effort has been put into studying the effects of tumors and their products on endothelial cells, particularly as it relates to tumor-induced angiogenesis. The classic examples are the many isoforms of VEGF, which are known to induce endothelial cell proliferation, chemotaxis, and enhancement of vessel permeability [40, 41]. Our own work has identified SEMA4D as a similar product of some malignancies that is chemotactic for endothelial cells and can enhance blood vessel content for the purpose of promoting tumor growth [4, 6]. However, only recently has a focus been placed on deciphering how malignancies affect mural cells such as pericytes, which regulate vessel integrity, maintenance, survival, and function [42]. Such work could have great significance for control of pathological angiogenesis, as attempting to inhibit pericyte association with endothelial cells along with targeting the endothelium itself is an emerging aspect of anti-angiogenic intervention in the treatment of cancer. Indeed, though tumor vessels are known to be heterogeneous in their pericyte coverage, there is evidence that anti-angiogenic therapy targeting VEGF/VEGFR-2 may lead to ablation of naked endothelial tubes only, while pericyte covered segments remain resistant, possibly contributing to treatment failure [43–45]. In an attempt to evaluate the effects of SEMA4D inhibition on tumor-induced angiogenesis and compare it to VEGF blockade, we noted that inhibition of both restricted tumor vascularity and size but vessels forming under conditions of VEGF blockade retained their association with pericytes while those arising in a background of SEMA4D/Plexin-B1 deficiency did not [8]. We therefore looked for possible mechanisms for how SEMA4D blockade would not only decrease vascular density but also affect pericyte coverage and vascular stability.
We have previously shown that many of the pro-angiogenic effects of Plexin-B1 signaling are mediated by the small GTPase RhoA, including activation of AKT and ERK [46]. While RhoA plays a key role in cell migration, adhesion, and stress fiber formation, it also regulates gene expression and nuclear signaling [47]. We noted that treatment of HUVEC with SEMA4D induced production of PDGF-B in a Plexin-B1/RhoA-dependent manner. PDGF-B is a crucial factor in the activation and recruitment of pericytes to newly formed vessels [11]. PDGF-B is normally expressed by sprouting capillary endothelial cells only at sites where active angiogenesis is taking place, whereas its receptor, PDGFR-β, is found on pericytes [48, 49]. We were interested in examining if tumor cells have the ability to differentiate and recruit pericytes when establishing new blood vessels via SEMA4D-mediated induction of PDGF-B, either from endothelium or possibly tumor cells themselves, as both endothelial and non-endothelial cells have been shown to recruit pericytes to tumor blood vessels through PDGF-B signaling networks [34, 50]. We demonstrated that SEMA4D from HNSCC cells exerted a small autrocrine effect on production of PDGF-B but more importantly seemed to induce its production from endothelial cells, explaining why we observed such a robust influence on pericyte proliferation and migration in media conditioned by SEMA4D treated HUVEC, and association of pericytes with HUVECs in co-culture, effects that could be inhibited by concurrent administration of PDGF-B blocking antibody or silencing of HUVEC Plexin-B1.
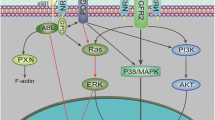
Pericytes are believed to arise at least in part by PDGF-B-induced differentiation of mesenchymal stem cells, which also have the potential to differentiate along an endothelial lineage in the presence of VEGF [12, 23, 51]. We showed that media conditioned by HUVECs treated with SEMA4D induced RGS5, NG2 and α-SMA expression in C3H/10T1/2 embryonic mesenchymal stem cells, markers of pericyte differentiation [24, 35]. The model we propose for SEMA4D and VEGF is illustrated in Fig. 5, and could explain our original observation (and that of others [10]) as to why inhibition of VEGF in tumors leaves the few remaining vessels with pericyte coverage: while VEGF inhibition prevents formation of new endothelial cells from stem cell precursors and enhances apoptosis of newly formed endothelial cells without pericyte protection (naked endothelial tubes), unaffected PDGF-B production induced in surviving endothelial cells by tumor-derived SEMA4D directs differentiation of mesenchymal stem cells toward a pericyte linage. The unintended consequence would be VEGF blockade indirectly driving differentiation of stem cells to pericytes [51], resulting in sheathed, stable vessels protected from further anti-angiogenic intervention, and attenuated effectiveness of anti-angiogenic therapy. In contrast, inhibition of SEMA4D also causes endothelial cell apoptosis and reduced vascular density [8, 9] but decreases available PDGF-B, leading to fewer pericytes. Vessels without pericyte sheaths are immature and may be more reliant on growth factors like VEGF for survival. Blocking SEMA4D could therefore render vessels more susceptible to destruction when combined with VEGF blockade. In fact, we have observed enhanced endothelial cell apoptosis and greatly reduced size of tumors when both SEMA4D and VEGF were inhibited compared to inhibition of either factor alone, and growth arrest even in anti-VEGF therapy-resistant neoplasms, suggesting that SEMA4D blockade could not only be an excellent form of treatment concurrent with anti-VEGF therapy but also when anti-VEGF therapy has failed [8, 9]. Anti-SEMA4D therapy also would likely be very safe, as Plexin-B1 signaling is redundant in normal vascular development [52].

Effects of tumor-derived SEMA4D and VEGF on vasculature. VEGF production by tumors drives stem cells towards endothelial cell differentiation, enhancing angiogenesis in the tumor stroma. In the absence of VEGF, for example during anti-VEGF therapy, tumor cell SEMA4D would predominate. SEMA4D favors stem cell differentiation toward pericytes via endothelial production of PDGF-B and induces endothelial production of ANGPTL4, which itself is pro-angiogenic and promotes vascular permeability. In the absence of SEMA4D endothelial cells would fail to make PDGF-B, thus reducing the number of pericytes, and the remaining vessels would be less permeable concurrent with a reduction in ANGPTL4
While investigating SEMA4D-mediated induction of PDGF-B from HUVEC, we also noted upregulation of ANGPTL4, another significant difference in the response of endothelial cells from that of VEGF. Previous studies have identified ANGPTL4 as a hypoxia-related gene, with endothelial cells exhibiting elevated ANGPTL4 mRNA and protein levels in response to low oxygen tension [18]. There is evidence that ANGPTL4 exerts a VEGF-independent proangiogenic effect, suggesting that it could be a key modulator in tumor angiogenesis, particularly in a hypoxic microenvironment caused by a rapidly growing malignancy. For example, ANGPTL4 is important in Kaposi Sarcoma, where its expression enhances endothelial cell migration and differentiation, both of which are important in angiogenesis and tumor vascularity [19]. Neovascularization also influences the metastatic spread of cancer cells throughout the body. Indeed, the vascularization levels of most solid tumors are thought to correlate with metastatic potential, with metastasis depending upon not only blood vessel density but also increased vasculature leakiness which is required for passage of tumor cells in and out of the vessels, a process influenced by ANGPTL4. Studies have shown that ANGPTL4 disrupts vascular endothelial cell–cell junctions by directly interacting with VE-cadherin, increases permeability of lung capillaries, and facilitates trans-endothelial passage of tumor cells, thereby promoting metastasis [53]. Mice deficient in ANGPTL4 exhibit reduced vascular permeability and attenuated lung metastasis [20]. Our results support these findings, as we could demonstrate SEMA4D-mediated induction of ANGPTL4 from HUVEC which served to induce cadherin internalization and presumably vascular permeability (Fig. 5), an effect that was lost upon silencing of ANGPTL4. We believe this would also result in enhanced metastasis of tumor xenografts, with disruption of SEMA4D/Plexin-B1/ANGPTL4 signaling suppressing this response, a possibility that we are currently investigating.
While treatments with VEGF inhibitors like Avastin have shown clinical benefit, more dramatic results might be achieved by combination therapy with other anti-angiogenic agents. It is known that SEMA4D, both soluble and derived from tumors, can promote angiogenesis in a VEGF-independent manner. Here we show that it not only does this directly, but it acts upon endothelial cells to recruit pericytes to these newly formed vessels, a process which serves to stabilize and protect vessels from anti-angiogenic intervention, and to induce ANGPTL4, which itself is pro-angiogenic and also promotes vascular permeability and metastasis. Combinations of anti-SEMA4D and anti-VEGF agents are likely to provide additional pruning of blood vessels and suppression of metastasis.
Change history
26 February 2020
The Editors-in-Chief have retracted this article [1] following an investigation by the University of Maryland. The institution found that in Figure 1C, the graph showing PDGF-B does not match the original data for the 24-hour time point. The graph shows the value to be over 1000 pg/ml, but the original data have a value of 106.626. In Figure 1F, the data were entered manually to create the standard deviation bars. The data manually entered do not match the original data. When the standard deviations for the original data were calculated, the p values were no longer significant using a paired student t test. In Figure 2C, the original data do not match the published data. In Figure 4B, the images in the first lane and the fifth lane are from the same micrograph (i.e., the same set of conditions). However, the published figure claims that they are different conditions. The metadata in this figure also shows different cell lines than those noted in the article. The first and last images are labelled as Du145 shAR3 anti AR3.jpg. The second image is labelled as Du145 shAR8 anti AR8.jpg. The third image is labelled as Cos1 mARs3 mS3-2 antibody-2.jpg. The fourth image is labelled as R1 3634 bleed.jpg. Due to these errors, the Editors-in-Chief have found that the results are no longer reliable.
26 February 2020
The Editors-in-Chief have retracted this article [1] following an investigation by the University of Maryland. The institution found that in Figure 1C, the graph showing PDGF-B does not match the original data for the 24-hour time point. The graph shows the value to be over 1000 pg/ml, but the original data have a value of 106.626. In Figure 1F, the data were entered manually to create the standard deviation bars. The data manually entered do not match the original data. When the standard deviations for the original data were calculated, the p values were no longer significant using a paired student t test. In Figure 2C, the original data do not match the published data. In Figure 4B, the images in the first lane and the fifth lane are from the same micrograph (i.e., the same set of conditions). However, the published figure claims that they are different conditions. The metadata in this figure also shows different cell lines than those noted in the article. The first and last images are labelled as ���Du145 shAR3 anti AR3.jpg���. The second image is labelled as ���Du145 shAR8 anti AR8.jpg���. The third image is labelled as ���Cos1 mARs3 mS3-2 antibody-2.jpg.��� The fourth image is labelled as ���R1 3634 bleed.jpg���. Due to these errors, the Editors-in-Chief have found that the results are no longer reliable.
References
Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, Tessier-Lavigne M, Comoglio PM (1999) Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99:71–80
Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M (1998) Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92:735–745
Hota PK, Buck M (2012) Plexin structures are coming: opportunities for multilevel investigations of semaphorin guidance receptors, their cell signaling mechanisms, and functions. Cell Mol Life Sci 69:3765–3805
Basile JR, Barac A, Zhu T, Guan KL, Gutkind JS (2004) Class IV semaphorins promote angiogenesis by stimulating Rho-initiated pathways through plexin-B. Cancer Res 64:5212–5224
Conrotto P, Valdembri D, Corso S, Serini G, Tamagnone L, Comoglio PM, Bussolino F, Giordano S (2005) Sema4D induces angiogenesis through Met recruitment by Plexin B1. Blood 105:4321–4329
Basile JR, Castilho RM, Williams VP, Gutkind JS (2006) Semaphorin 4D provides a link between axon guidance processes and tumor-induced angiogenesis. PNAS 103:9017–9022
Sun Q, Zhou H, Binmadi NO, Basile JR (2009) Hypoxia-inducible factor-1-mediated regulation of semaphorin 4D affects tumor growth and vascularity. J Biol Chem 284:32066–32074
Zhou H, Binmadi NO, Yang YH, Proia P, Basile JR (2012) Semaphorin 4D cooperates with VEGF to promote angiogenesis and tumor progression. Angiogenesis 15:391–407
Zhou H, Yang YH, Binmadi NO, Proia P, Basile JR (2012) The hypoxia-inducible factor-responsive proteins semaphorin 4D and vascular endothelial growth factor promote tumor growth and angiogenesis in oral squamous cell carcinoma. Exp Cell Res 18:1685–1698
Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, Ross S, Cheng ZY, Le Couter J, Plowman G, Peale F, Koch AW, Wu Y, Bagri A, Tessier-Lavigne M, Watts RJ (2007) Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell 11:53–67
Betsholtz C (2004) Insight into the physiological functions of PDGF through genetic studies in mice. Cytokine Growth Factor Rev 15:215–228
Bergers G, Song S (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 7:452–464
Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D (2003) Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest 111:1287–1295
Ge H, Yang G, Huang L, Motola DL, Pourbahrami T, Li C (2004) Oligomerization and regulated proteolytic processing of angiopoietin-like protein 4. J Biol Chem 279:2038–2045
Mandard S, Zandbergen F, Tan NS, Escher P, Patsouris D, Koenig W, Kleemann R, Bakker A, Veenman F, Wahli W, Muller M, Kersten S (2004) The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. J Biol Chem 279:34411–34420
Hu Z, Fan C, Livasy C, He X, Oh DS, Ewend MG, Carey LA, Subramanian S, West R, Ikpatt F, Olopade OI, van de Rijn M, Perou CM (2009) A compact VEGF signature associated with distant metastases and poor outcomes. BMC Med 7:9
Nakayama T, Hirakawa H, Shibata K, Nazneen A, Abe K, Nagayasu T, Taguchi T (2011) Expression of angiopoietin-like 4 (ANGPTL4) in human colorectal cancer: ANGPTL4 promotes venous invasion and distant metastasis. Oncol Rep 25:929–935
Le Jan S, Amy C, Cazes A, Monnot C, Lamande N, Favier J, Philippe J, Sibony M, Gasc JM, Corvol P, Germain S (2003) Angiopoietin-like 4 is a proangiogenic factor produced during ischemia and in conventional renal cell carcinoma. Am J Pathol 162:1521–1528
Ma T, Jham BC, Hu J, Friedman ER, Basile JR, Molinolo A, Sodhi A, Montaner S (2010) Viral G protein-coupled receptor up-regulates Angiopoietin-like 4 promoting angiogenesis and vascular permeability in Kaposi’s sarcoma. Proc Natl Acad Sci U S A 107:14363–14368
Huang RL, Teo Z, Chong HC, Zhu P, Tan MJ, Tan CK, Lam CR, Sng MK, Leong DT, Tan SM, Kersten S, Ding JL, Li HY, Tan NS (2011) ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 clusters. Blood 118:3990–4002
Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J (2005) Genes that mediate breast cancer metastasis to lung. Nature 436:518–524
Cardinali M, Pietraszkiewicz H, Ensley JF, Robbins KC (1995) Tyrosine phosphorylation as a marker for aberrantly regulated growth-promoting pathways in cell lines derived from head and neck malignancies. Int J Cancer 61:98–103
Dhar K, Dhar G, Majumder M, Haque I, Mehta S, Van Veldhuizen PJ, Banerjee SK, Banerjee S (2010) Tumor cell-derived PDGF-B potentiates mouse mesenchymal stem cells-pericytes transition and recruitment through an interaction with NRP-1. Mol Cancer 9:209
Darland DC, D’Amore PA (2001) TGF beta is required for the formation of capillary-like structures in three-dimensional cocultures of 10T1/2 and endothelial cells. Angiogenesis 4:11–20
Hannon GJ, Conklin DS (2004) RNA interference by short hairpin RNAs expressed in vertebrate cells. Methods Mol Biol 257:255–266
Siolas D, Lerner C, Burchard J, Ge W, Linsley PS, Paddison PJ, Hannon GJ, Cleary MA (2005) Synthetic shRNAs as potent RNAi triggers. Nat Biotechnol 23:227–231
Yang YH, Zhou H, Binmadi NO, Proia P, Basile JR (2011) Plexin-B1 activates NF-kappaB and IL-8 to promote a pro-angiogenic response in endothelial cells. PLoS ONE 6:e25826
Paddison PJ, Caudy AA, Sachidanandam R, Hannon GJ (2004) Short hairpin activated gene silencing in mammalian cells. Methods Mol Biol 265:85–100
Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O’Shaughnessy A, Gnoj L, Scobie K, Chang K, Westbrook T, Cleary M, Sachidanandam R, McCombie WR, Elledge SJ, Hannon GJ (2004) A resource for large-scale RNA-interference-based screens in mammals. Nature 428:427–431
Franco M, Roswall P, Cortez E, Hanahan D, Pietras K (2011) Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood 118:2906–2917
Amornphimoltham P, Sriuranpong V, Patel V, Benavides F, Conti CJ, Sauk J, Sausville EA, Molinolo AA, Gutkind JS (2004) Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res 10:4029–4037
Perrot V, Vazquez-Prado J, Gutkind JS (2002) Plexin B regulates Rho through the guanine nucleotide exchange factors leukemia-associated Rho GEF (LARG) and PDZ-RhoGEF. J Biol Chem 277:43115–43120
Aurandt J, Vikis HG, Gutkind JS, Ahn N, Guan KL (2002) The semaphorin receptor plexin-B1 signals through a direct interaction with the Rho-specific nucleotide exchange factor, LARG. Proc Natl Acad Sci U S A 99:12085–12090
Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landegren U, Nystrom HC, Bergstrom G, Dejana E, Ostman A, Lindahl P, Betsholtz C (2003) Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev 17:1835–1840
Berger M, Bergers G, Arnold B, Hammerling GJ, Ganss R (2005) Regulator of G-protein signaling-5 induction in pericytes coincides with active vessel remodeling during neovascularization. Blood 105:1094–1101
Elhabazi A, Delaire S, Bensussan A, Boumsell L, Bismuth G (2001) Biological activity of soluble CD100. I. The extracellular region of CD100 is released from the surface of T lymphocytes by regulated proteolysis. J Immunol 166:4341–4347
Basile JR, Holmbeck K, Bugge TH, Gutkind JS (2007) MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J Biol Chem 282:6899–6905
Binmadi NO, Yang YH, Zhou H, Proia P, Lin YL, Batista De Paula AM, Sena Guimaraes AL, Poswar FO, Sundararajan D, Basile JR (2012) Plexin-B1 and Semaphorin 4D cooperate to promote perineural invasion in a RhoA/ROK-dependent manner. Am J Pathol 180:1232–1242
Zhu P, Goh YY, Chin HF, Kersten S, Tan NS (2012) Angiopoietin-like 4: a decade of research. Biosci Rep 32:211–219
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3:401–410
Gavard J, Gutkind JS (2006) VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 8:1223–1234
Abramsson A, Lindblom P, Betsholtz C (2003) Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest 112:1142–1151
Baluk P, Hashizume H, McDonald DM (2005) Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15:102–111
Erber R, Thurnher A, Katsen AD, Groth G, Kerger H, Hammes HP, Menger MD, Ullrich A, Vajkoczy P (2004) Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J 18:338–340
Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E (1999) Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 103:159–165
Basile JR, Gavard J, Gutkind JS (2007) Plexin-B1 utilizes RhoA and Rho kinase to promote the integrin-dependent activation of Akt and ERK and endothelial cell motility. J Biol Chem 282:34888–34895
Marinissen MJ, Gutkind JS (2001) G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 22:368–376
Armulik A, Abramsson A, Betsholtz C (2005) Endothelial/pericyte interactions. Circ Res 97:512–523
Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C (2001) Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 153:543–553
Guo P, Hu B, Gu W, Xu L, Wang D, Huang HJ, Cavenee WK, Cheng SY (2003) Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am J Pathol 162:1083–1093
Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, Naito M, Nakao K (2000) Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 408:92–96
Fazzari P, Penachioni J, Gianola S, Rossi F, Eickholt BJ, Maina F, Alexopoulou L, Sottile A, Comoglio PM, Flavell RA, Tamagnone L (2007) Plexin-B1 plays a redundant role during mouse development and in tumour angiogenesis. BMC Dev Biol 7:55
Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, Massague J (2008) TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133:66–77
Acknowledgments
We would like to acknowledge Dr. Snigdha Banerjee for the use of C3H/10T1/2 embryonic mesenchymal stem cells. This work was supported by the National Cancer Institute grant R01-CA133162 to J.R.B.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
The Editors-in-Chief have retracted this article following an investigation by the University of Maryland. The institution found that problems in some figures. Due to these errors, the Editors-in-Chief have found that the results are no longer reliable.
About this article
Cite this article
Zhou, H., Yang, YH. & Basile, J.R. RETRACTED ARTICLE: The Semaphorin 4D-Plexin-B1-RhoA signaling axis recruits pericytes and regulates vascular permeability through endothelial production of PDGF-B and ANGPTL4. Angiogenesis 17, 261–274 (2014). https://doi.org/10.1007/s10456-013-9395-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-013-9395-0