
Abstract
Environmental samples analyzed by quantitative PCR (qPCR) are subject to interference by inhibitors present in the environment being sampled. A controlled determination of the effect of inhibitors associated with sampling indoor air and the ability of internal standard controls to detect inhibition was carried out by filter collection of air samples followed by spiking of the filters with green fluorescent protein-expressing Aspergillus fumigatus conidia. Microscopic conidial counts were compared with qPCR results and correlated with levels of particulate matter and viable airborne microorganisms. Our data showed that PCR can be inhibited by masses of particulate matter as low as 50 μg and that the amount of inhibition was positively correlated with the mass of particulate (r = 0.75) and the number of non-filamentous organisms (r = 0.73). The use of internal standard DNA identified the presence of inhibitors and indicated the need for additional sample processing or qualification of sample results.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The principal route of exposure to fungi is via inhalation of airborne viable and non-viable fungal particles. Some viable fungal spores have the capacity to establish invasive infections, cause hypersensitivity diseases, elicit toxic effects, and produce volatile organic compounds (Burge, Pierson, Groves, Strawn, & Mishra, 2000). Exposure assessment professionals are often faced with the daunting task of rapidly detecting very low numbers of organisms, such as fungi, in environmental samples. Assessing exposure to airborne fungi is difficult because there are no accepted standard sampling protocols or exposure guidelines. Historically, air samples have been analyzed by direct microscopic examination of collected spores or, more commonly, by growing collected viable spores on an appropriate culture medium under suitable growth conditions (Burge et al., 2000; Griffiths & DeCosemo, 1994). Major limitations preventing the utility of these classical techniques include sample size and time limitations, inaccurate characterization of isolated microbes, overloading of collection media, and lengthy analysis times (McDevitt, Lees, Merz, & Schwab, 2005). Industrial hygiene practitioners have recently begun to evaluate the utility of molecular detection methods to overcome the constraints of conventional analysis. In particular, the PCR has shown great promise. Over the last decade, quantitative PCR (qPCR) has begun to replace basic PCR. The major advantage of the former molecular technique is its capacity to enumerate the target template, thus providing more information than a simple qualitative yes/no result. The utility of qPCR for quantitative measurement of potentially pathogenic fungi, such as Aspergillus fumigatus, has been demonstrated and reported previously (Costa et al., 2001; Haugland, Vesper, & Wymer, 1999; McDevitt, Lees, Merz, & Schwab, 2004, 2005; Roe, Haugland, Vesper, & Wymer, 2001; Spiess et al., 2003; Zhou, Whong, Ong, & Chen, 2000). qPCR analysis of A. fumigatus and other fungal conidia DNA is sensitive and specific, has a quantitative linear response (when data are transformed from an exponential reaction) over many orders of magnitude, and can provide results within a few hours (Costa et al., 2001; Cruz-Perez, Buttner, & Stetzenbach, 2001; McDevitt et al., 2004, 2005). The use of TaqMan probes as a part of qPCR analyses facilitates the continuous monitoring of PCR amplification curves by measuring the signal generated when a fluorescently labeled probe annealed to a target nucleic acid sequence is cleaved during polymerase extension (Livak, Flood, Marmaro, Giusti, & Deetz, 1995). The fluorescent probe contains a specific fluor and corresponding fluor quencher at the 5′ and 3′ ends of the probe, respectively. Cleavage of the probe by Taq polymerase liberates the fluor from the quencher, thereby allowing the fluor to be measured by the detector. The fractional cycle during PCR at which the fluorescent signal is resolved from a designated threshold (detection threshold) above baseline is called the threshold cycle (C t). The greater the initial number of target templates, the fewer amplification cycles are required to exceed the threshold. There is a linear (negative slope) relationship between C t and log10 of initial template number. Thus, low C t values are associated with high initial copy numbers, and high C t values are associated with low initial copy numbers. The C t determined for unknown samples can be compared to a standard curve constructed using C t values determined for known amounts of template nucleic acid.
A major limitation encountered when using qPCR for environmental sample analyses is the presence of exogenous substances, called inhibitors, which are commonly found in environmental samples. These inhibitors can interfere with the PCR amplification reaction and yield C t values which do not reflect the actual concentrations of target present in a sample (McDevitt et al., 2004; Wilson, 1997). The problem of inhibition has been recognized for some time in many areas of research relying on PCR analysis of nucleic acid (Alvarez, Buttner, & Stetzenbach, 1995; Wilson, 1997). Inhibitors can cause the complete or partial failure of the PCR reaction. Complete inhibition, when undetected, may yield false negative results since nucleic acid is not detected even when present, while partial inhibition may lead to underreporting of nucleic acid concentrations due to a lowered PCR signal (i.e., increased C t values). Inhibitors may have many sources, interfere with the PCR in a variety of ways, and can have complex interactions that make the identification of inhibition problematic. The source of the sample containing the target microbe of interest, such as clinical, food, or environmental (air, water, soil, etc.) sources, will greatly affect the potential for inhibition and the type of inhibitors present. Wilson (1997) provides an extensive list of sample substrates and associated inhibitors. Trace amounts of humic substances, heavy metals, constituents of bacterial cells, proteolytic enzymes, and high concentrations of non-target DNA have all been associated with the inhibition of the PCR in environmental matrices such as air, soil, water, sewage, and sediment (Alvarez et al., 1994, 1995; Keswani, Kashon, & Chen, 2005; Maher, Dillon, Vermund, & Unnasch, 2001; Roe et al., 2001; Wilson, 1997).
The addition of an internal standard control, such as exogenous DNA or reference organisms, has been used to detect and identify the presence of PCR inhibitors (Courtney, Smith, & Henchal, 1999; McDevitt et al., 2004; Sachadyn & Kur, 1998; Schwab, Neill, Le Guyader, Estes, & Atmar, 2001). Internal standard DNA controls are designed to be amplified in a manner similar to target DNA, while being sufficiently unique to allow discrete differentiation between the target and internal standard DNA. Ideal internal controls have a similar length and base-pair content as the target sequence, yet have a unique primer and/or probe sequence (Courtney et al., 1999; Sachadyn & Kur, 1998). The detection of the PCR control DNA at the anticipated C t value in the absence of target DNA generally shows that the amplification conditions were appropriate and that the target DNA was not present. Likewise, a reduction or change in the expected C t values of the internal standard in a sample compared to those of the internal standard DNA control samples suggests that partial inhibition has occurred and that the signals from the target DNA may also be reduced.
We have previously developed a qPCR method optimized for environmental sampling for A. fumigatus conidia that incorporated: single tube sample preparation to reduce sample loss, maintain simplicity, and avoid contamination; hot start amplification to reduce non-specific primer/probe annealing; uracil-N-glycosylase to prevent carryover contamination (McDevitt et al., 2004). Using this method, we demonstrated that with the addition of internal standard control DNA to the PCR master mix we were able to detect complete and partial sample inhibition of A. fumigatus DNA amplification caused by inhibitors spiked into qPCR reaction mixes. Additional studies evaluated the use of green fluorescent protein (GFP)-expressing A. fumigatus conidia to compare qPCR enumeration with direct epifluorescent microscopic filter counts of GFP-containing conidia (McDevitt et al., 2005). We showed that the qPCR analysis method was in agreement (1:1 ratio between qPCR and direct microscopic estimates of filter loading ranging from 15–30,000 conidia per filter) with an external reference microscopy method and, therefore, can be considered to be a reliable method for enumerating A. fumigatus conidia collected on filters.
The experiments described above were performed in a highly controlled environment within a laboratory aerosol test chamber and did not reflect variables associated with actual environmental sampling. The goals of the present study were to extend our evaluation of qPCR detection of fungi in a controlled fashion and to add the reality of background airborne particulate matter (PM) that would be found during environmental sampling by incorporating environmental sampling to collect naturally occurring PM onto filters. We subsequently seeded the filters with GFP-expressing A. fumigatus conidia to facilitate our use of qPCR analysis along with direct microscopic counting as previously described. The extent of inhibition was quantitatively analyzed through the use of internal standard DNA and comparison of qPCR estimates of conidia number with optical counts. In order to analyze the frequency, extent, and causes of inhibition of qPCR, we characterized the amount of PM collected and estimated the concentration of bioaerosols present in the environment during the sampling period.
2 Methods
2.1 Sampling plan
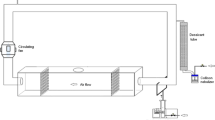
Sampling was conducted during a single 8-h interval (between 7:30 a.m. and 6:30 p.m.) at three locations in Baltimore, Maryland during the month of November 2003: an office, an office with documented indoor air quality complaints, and a home. A schematic representation of the sampling and analysis plan is shown in Fig. 1. The plan was designed to provide varying amounts of PM from each sampling location by varying the sample collection time.

Schematic diagram of sampling and analysis plan for field sampling at each site
2.2 PM collection
At each sampling location, air was drawn through three sets of two Isopore filters (diameter: 25 mm; pore size: 0.8 μm; Millipore, Bedford, Mass.) in open-faced cassettes at a flow rate of approximately 10 l per minute (lpm) using three high-volume sampling pumps. Each sampling pump was attached to two filters via a Y-connection (Fig. 2). A precision rotometer was placed in each arm of the Y to control and adjust the flow to each filter cassette. The rotometers were calibrated (within the sampling train) against a Dry Cal DC-2 Flow Calibrator (Bios International, Butler, NY). All filters were located in the same proximity to each other (Fig. 2). All high-volume pumps were turned on at the same time. Pump 1 was turned off after 120 min, pump 2 was turned off after 240 min, and pump 3 was turned off after operating for 480 min.

Field sampling monitoring equipment set-up. A Filters, B rotometers, C MIE real-time particle sampler, D Anderson N-6 sampler, E air sampling pumps
2.3 Gravimetric analysis
Since the amount of PM collected on the filter may be correlated with sample inhibition, the filters were weighed before and after sampling to determine the amount of PM collected during sampling, as described in the preceding section. Prior to weighing, numbered filter cassettes (without plugs) were placed in a desiccator for 48 h. The filters were then removed from the cassette and weighed two consecutive times in an environmentally controlled room (21 ± 3°C and 40 ± 5% relative humidity) using a Mettler MT5 microbalance (Mettler, Toledo, Ohio) and the filter number and masses recorded. After sampling, each filter was desiccated for 48 h, reweighed two consecutive times, and the filter number and mass recorded.
2.4 Viable samples
An N-6 Single Stage Viable Impactor (Thermo Electron, Cheswick, Pa.) was used to collect viable bioaerosol samples directly onto culture plates during field sampling. Replicate samples were collected on 100-mm disposable petri dishes (Fisher Scientific, Norcross, Ga.) containing 39 ml of Sabouraud dextrose agar (Becton, Dickson and Company, Sparks, Md.). To prevent cross-contamination prior to sampling, the N-6 sampler was first swabbed with 70% ethanol and then the pump was turned on for about 60 s to evaporate the ethanol. Viable samples were collected at the following times with respect to filter sampling: 0 min, +120 min, +240 min, and +480 min. Each sample was collected over a 4-min period at a flow rate of 28.3 lpm. The flow rate was checked before and after sampling at each site using a rotometer, which was calibrated against a Dry Cal DC-2 Flow Calibrator. The plates were stored at 4°C after each sampling interval. After the last sample was collected for the day, the plates were incubated at 25°C. The plates were checked for growth at 24-h intervals, and the number of filamentous and non-filamentous colony forming units (CFU) was recorded. Any colonies with gross morphology consistent with Aspergillus species of fungi were transferred to potato dextrose agar plates for identification based on colony and microscopic morphological characteristics.
2.5 Real-time particle concentration monitoring
Real-time aerosol concentrations were monitored during field sampling using the MIE personalDataRAM model pDR-100AN (Thermo Andersen, Smyrna, Ga.). The pDR100AN, which operates as a passive sampler, was factory-calibrated, zeroed prior to sampling, and set to a 60-s logging interval. Monitoring was performed using the pDR100AN for the entire 8-h sampling period. After sampling was completed, stored data were downloaded to a computer using the MIE software package.
2.6 Seeding and analysis of GFP conidia on filters
Following environmental sampling, GFP-containing A. fumigatus conidia (Wasylnka & Moore, 2002) which are 2–3 μm in diameter, were seeded onto a subset of the filters used to collect particulate matter (as outlined in the section on ‘PM collection’). Suspensions of GFP A. fumigatus were aerosolized using a Collison nebulizer (BGI, Waltham, Mass.) and introduced into a polycarbonate chamber equipped with a mixing fan, as previously described (McDevitt et al., 2005). Briefly, chamber air was drawn through an open-face filter cassette at 3 l per minute for 5 min. After sample collection in the aerosol chamber, each filter was cut in half for a comparison of methods. One-half was analyzed by qPCR (described below), and the remaining half was enumerated by directly counting conidia on representative areas of the filter surface using epifluorescence microscopy at 400× total power, as described previously (McDevitt et al., 2005). qPCR analysis involved placing the seeded filter half into a 1.5-ml centrifuge tube containing 150 μl of lyticase buffer solution (200 U lyticase, 1.6 mM Tris, 0.03 μM EDTA, pH 7.5) and agitating the tube in a mini-beadbeater (without beads) at 5000 rpm for 30 s. After agitation, the filter was spun dry at 23,000g for 5 min, removed and discarded; the conidia digest was then incubated for 60 min at 25°C, agitated with the mini-beadbeater for 30 s at 5000 rpm, and stored at 4°C until qPCR analysis.
TaqMan qPCR was performed using the forward primer (AF7) 5′-GAA AGG TCA GGT GTT CGA GTC A-3′, the reverse primer (AF8) 5′-CAT CAT GAG TGG TCC GCT TTA C-3′, and probe (AF9) 5′-FAM-ATC CCT AAA CCC GCA ACC AAA GGC- BHQ-1-3′, all of which were designed to amplify a fragment of the nine-to-ten copy A. fumigatus mitochondrial gene described by Costa et al. (2001). Primers were purchased from Invitrogen Custom Primers (Frederick, Md.), and probes were purchased from Biosearch Technologies (Novato, Calif.). The qPCR amplification of 10 μl of conidia digest was performed in a 25-μl total volume reaction mixture containing 1.5 U AmpliTaq Gold polymerase (Applied Biosystems, Foster City, Calif.), 1× Gene Amp PCR Buffer II (Applied Biosystems), 3.0 μM MgCl2 (Applied Biosystems), 0.5 U Amperase Uracil N-glycosylase (UNG) (Applied Biosystems), 200 μM dATP, dCTP, dGTP/400 μM dUTP (Promega, Madison, Wis.), 0.2 μM AF7 and AF8 primers, and 0.2 μM AF9 probe. PCR amplification and quantitative analysis were performed in a Smart Cycler (Cepheid, Sunnyvale, Calif.) and consisted of an initial UNG digestion period (2 min at 50°C), an AmpliTaq Gold activation interval (10 min at 95°C), followed by 50 cycles of repeated denaturation (15 s at 95°C) and annealing/extension (60 s at 65°C). Threshold concentration (C t) analysis was performed using Smart Cycler software using second-derivative curve analysis.
3 Results
The average airborne concentration of PM measured over the 8-h sampling period at each location is shown for real time measurements with the MIE and gravimetric analysis in Fig. 3. Based on MIE measurements, the 8-h average PM concentrations were generally similar between the three sites. The highest 8-h average was noted at site 2, and the highest peak concentrations (data not shown) were measured at site 3. However, the 8-h average gravimetric results for particulate sampling on filters (Fig. 3) revealed that the average room air particulate concentrations were markedly higher at site 3. The mass of PM measured by gravimetric analysis for each filter sample is shown in Fig. 4. As expected, there is a general increase in mass associated with sampling time. The mass loadings at sampling sites 1 and 2 are similar, while the trends in corresponding samples at site 3 reflect higher mass loadings.

Comparison of a single-day, 8-h average airborne particulate concentrations (n = 3) at sites 1, 2, and 3 as determined by MIE and gravimetric sampling

Comparison of average particulate matter (PM) mass collected for duplicate samples on a single day at field monitoring sites 1, 2, and 3
Airborne concentrations of viable filamentous fungi ranged from 20 to 1240 CFU/m3, as measured by the N-6 sampler for the four samples taken over the 8-h interval, while the airborne concentrations of other viable microorganisms ranged from <10 to 1060 CFU/m3. Based on the averages for the four viable samples collected during the 8-hour particulate sampling interval at each site (Fig. 5), the concentration of filamentous fungi at sites 1 and 3 were similar, and higher, than the concentration at site 2, while for non-filamentous organisms, the average numbers were relatively low at sites 1 and 2 and higher at site 3. The total numbers of filamentous and non-filamentous organisms collected for each filter are shown in Table 1. These values are based on the volume of air drawn through the filter and the estimated airborne concentrations of organisms (determined using the average viable sample results corresponding to each interval). Based on colony morphology and the inability of the non-filamentous colonies to grow on media containing antibiotics (data not shown), we assume these colonies to be bacteria.

Summary of 8-h average viable organism concentrations at sampling sites 1, 2, and 3
Aspergillus fumigatus was not detected by qPCR in any of the replicate samples analyzed to evaluate the level of endogenous A. fumigatus (i.e., background “A filters”) potentially present at the field sampling locations (data not shown). Both the limit of detection and the limit of quantification for these samples were 15 conidia per filter. Inhibition of qPCR was detected by the internal standard in all background samples from sites 2 and 3 (Table 1) when these samples were analyzed by qPCR. The detection of internal standard DNA was wholly inhibited in three of these samples (site 2—480 min, site 3—240 min, and site 3—480 min) and partially inhibited in three samples (site 2—120 min, site 2—240 min, and site 3—120 min). There was a positive correlation between levels of inhibition and mass of the PM (r = 0.75) and the number of non-filamentous organisms (r = 0.73). The mass of the PM and the number of non-filamentous organisms were also positively correlated (r = 0.94).
Table 2 presents a summary of the results of the qPCR analysis and direct microscopic counting as well as the results of the gravimetric analysis on the field samples that were seeded with GFP-A. fumigatus in the aerosol test chamber. The limit of detection and the limit of quantification for these samples were two conidia per filter half. A. fumigatus conidia were quantified by qPCR for all samples with the exception of site 3 (Table 2). All samples from site 3 were completely inhibited, some samples from site 2 were partially inhibited, and inhibition was not detected in any samples from site 1 when they were analyzed by qPCR with internal standard DNA. As noted previously, inhibition was associated with filters loaded with relatively higher particulate mass as compared to the PM mass of samples without inhibition (Table 1).
The estimated number of conidia per filter-half of the seeded field samples which were not completely inhibited ranged from 1898 to 3448 conidia per filter-half based on qPCR enumeration, and from 2142 to 4823 conidia per filter-half based on direct microscopic counts (Table 2). A comparison of these estimates showed that qPCR estimates of conidia counts were generally lower than those from direct microscopic counting.
4 Discussion
Our data demonstrates that PCR can be inhibited by masses of PM as low as 50 μg and that the amount of inhibition was positively correlated with the mass of particulate. This finding has serious implications, as it would not be uncommon to encounter masses of PM this magnitude during environmental air sampling. When one considers that the mean concentration of PM matter in Baltimore indoor air has been reported to be 56.5 μg/m3 (Breysse et al., 2005), an 8-h sample collected at a 10 lpm flow rate would result in the collection of 271 μg of PM. This amount of PM resulted in complete PCR inhibition in our study. It is obvious that one could decrease sampling times or flow rates to minimize the amount of PM collected; however, this would also result in a decrease in method sensitivity since fewer conidia would be collected under such conditions.
As noted previously, the causes of inhibition vary widely. Similarly, the composition of PM in the environment also varies a great deal with respect to concentration, particle size, and composition. As a result, identifying the specific component of PM which is inhibiting the PCR reaction would require characterization of the PM in terms of mass, size, and chemical composition. Since concentrations and composition of PM vary greatly both temporally and spatially, this characterization would have to be done for each sample. Although this strategy would be optimal, it would also be very costly and was beyond the scope of this study; additionally, it could not realistically be considered to be a part of routine analyses. However, we were able to characterize the PM collected in terms of viable organisms to a limited extent. We observed a positive correlation between PCR inhibition detected by internal standard DNA and the estimated number of viable non-filamentous organisms (assumed to be bacteria) collected per filter. We also noted a high correlation between the number of non-filamentous organisms and mass. Due to the small size and mass of bacteria, it is unlikely that the bacteria accounted for a large percentage of the mass of PM. Although significant numbers of bacteria were present in the inhibited samples, the relationship between samples with inhibition and PM and/or non-filamentous organisms was not resolved. Bacterial cells have been associated with PCR inhibition by other investigators (Alvarez et al., 1994; Olive, 1989). The proteases and nucleases associated with the bacteria may have caused physiochemical or enzymatic effects which interfered with amplification (Wilson, 1997).
Real-time monitoring was not necessarily representative of actual gravimetric sampling results and did not provide a reliable estimate of filter loading. Although the 8-h time-weighted average estimates of PM concentrations are in agreement with measurements by the MIE for field sampling sites 1 and 2, the amount of PM actually measured on the filter at field sampling site 3 was much greater than the amount that would have been predicted by the real-time measurement provided by the MIE (Fig. 3). This is not unexpected, since conditions at site 3 (residential) differed from those at sites 1 and 2 (offices). For example, there were two dogs and one cat present during sampling at site 3, while there was only minimal pedestrian traffic at sites 1 and 2. It would be reasonable to assume that particle size contributed to the differences noted between these methods. Microscopic evaluation of samples from site 3 showed many particles which exceeded the 10 μm upper particle size of maximum response for the MIE (data not shown). Since mass is approximately a cubed function of diameter, a small number of particles greater than 10 μm could account for a large proportion of the overall mass. Therefore, some of the most massive particles may have been excluded from the MIE measurements, resulting in this differential. The agreement between gravimetric analysis data and MIE data at sites 1 and 2 suggests that few particles larger than 10 μm were present at these locations. The activities of pets and lack of daily professional cleaning at site 3 may have contributed to the suspension of particles of larger diameters, thereby resulting in higher gravimetric results. The conditions at site 3 may have also been responsible for the differences in viable sampling results noted between site 3 and sites 1 and 2. Since these large particles do not efficiently penetrate the respiratory tract they are most likely not relevant to pulmonary pathology. As a result, the use of a size-selective sampler which excludes particles greater than 10 μm will significantly reduce the mass of particles collected and may yield a decrease in inhibition.
Investigators have reported the presence of inhibition in air and surface dust samples, but have provided minimal information regarding the nature and extent of this inhibition. Roe et al. (2001) reported a lower than expected PCR signal when Stachybotrys chartarum spores were seeded into milligram quantities of dust collected in heating, ventilation and air conditioning (HVAC) systems and from surfaces. Keswani et al. (2005) showed that inhibition varied with the type and amount of dust and the number of A. fumigatus or S. chartarum conidia that were seeded into surface dust and HVAC dust samples. The high amount of dust used in these experiments is not likely to be encountered when performing air sampling of indoor environments, such as homes and offices, which are expected to be relatively clean. During an investigation on Pneuomocystis carinii, Maher et al. (2001) noted a significant inhibition of PCR with filters used to collect outdoor air samples and a slight inhibition of PCR for indoor samples. Alvarez et al. (1995) report a complete inhibition associated with freeze-thaw cell lysis following the collection of environmental samples averaging 103–104 CFU/m3. However, these studies did not quantify the amount of inhibition nor attempt to associate the inhibition with sources.
The use of an internal standard DNA effectively detected instances in which the amplification of conidia DNA was totally inhibited (Table 2). There was only one instance of partial inhibition in these samples. The amount of inhibition detected by the internal standard DNA slightly exceeded the threshold of one C t unit change, while the numbers of conidia measured by qPCR were within expected limits. To show the presence and effect of potential inhibitors, our evaluation methods purposefully did not include the use of DNA purification methods to remove inhibitors. We had observed that the use of commercially available clean-up kits were marginally successful in removing the inhibition caused by airborne PM and resulted in greater than a log loss of PCR signal when tested with pure DNA without inhibitors present (data not shown).
Validation using optical counting was not an effective tool when high levels of PM were present of the filter. The field of view became more obscured as the amount of PM increased, and identification of conidia became increasingly difficult. At particulate loads above 100 μg per filter-half, accurate counting was not possible. Based on these findings, other means of validation are necessary when PM loads increase to greater than 100 μg per half-filter.
To our knowledge, this is the first report to examine the effect of airborne inhibitors of qPCR analysis in a controlled environment. Because of the inability to reliably predict the levels of A. fumigatus in the ambient air, we have collected naturally occurring PM onto filters and seeded the filters with GFP-expressing A. fumigatus conidia to facilitate the use of qPCR analysis along with direct microscopic counting with the aim of evaluating the role of inhibition in the assessment of inhalation exposure in indoor environments. We have shown that even modest amounts (≤50 μg) of PM found in indoor air environments can cause partial and/or complete inhibition of PCR reactions and that increasing PM mass correlates positively with the occurrence of inhibition. The use of internal standard DNA can identify the presence of inhibitors and the need for additional sample processing or qualification of sample results. PCR is a powerful tool for identifying and quantifying nucleic acid associated with organisms present in the environment; however, investigators must be aware of and control the limitations of PCR technology.
References
Alvarez, A. J., Buttner, M. P., Toranzos, G. A., Dvorsky, E. A., Toro, A., Heikes, T. B., Mertikas-Pifer, L. E., & Stetzenbach, L. D. (1994). Use of solid-phase PCR for enhanced detection of airborne microorganisms. Applied and Environmental Microbiology, 60, 374–376.
Alvarez, A. J., Buttner, M. P., & Stetzenbach, L. D. (1995). PCR for bioaerosol monitoring: Sensitivity and environmental interference. Applied and Environmental Microbiology, 61, 3639–3644.
Breysse, P. N., Buckley, T. J., Williams, D., Beck, C. M., Jo, S. J., Merriman, B., Kanchanaraksa, S., Swartz, L. J., Callahan, K. A., Butz, A. M., Rand, C. S., Diette, G. B., Krishnan, J. A., Moseley, A. M., Curtin-Brosnan, J., Durkin, N. B., & Eggleston, P. A. (2005). Indoor exposures to air pollutants and allergens in the homes of asthmatic children in inner-city Baltimore. Environmental Research, 98, 167–176.
Burge, H. A., Pierson, D. L., Groves T. O., Strawn K. F., & Mishra S. K. (2000). Dynamics of airborne fungal populations in a large office building. Current Microbiology, 40, 10–16.
Costa, C., Vidaud, D., Olivi, M., Bart-Delabesse, E., Vidaud, M., & Bretagne, S. (2001). Development of two real-time quantitative TaqMan PCR assays to detect circulating Aspergillus fumigatus DNA in serum. Journal of Microbiological Methods, 44, 263–269.
Courtney, B. C., Smith, M. M., & Henchal, E. A. (1999). Development of internal controls for probe-based nucleic acid diagnostic assays. Analytical Biochemistry, 270, 249–256.
Cruz-Perez, P., Buttner, M. P., & Stetzenbach, L. D. (2001). Detection and quantitation of Aspergillus fumigatus in pure culture using polymerase chain reaction. Molecular and Cellular Probes, 15, 81–88.
Griffiths, W. D., & DeCosemo, G. A. L. (1994). The assessment of bioaerosols – a critical review. Journal of Aerosol Science, 25, 1425–1458.
Haugland, R. A., Vesper, S. J., & Wymer, L. J. (1999). Quantitative measurement of Stachybotrys chartarum conidia using real time detection of PCR products with the TaqMan fluorogenic probe system. Molecular and Cellular Probes, 13, 329–340.
Keswani, J., Kashon, M. L., & Chen, B. T. (2005). Evaluation of interference to conventional and real-time PCR for detection and quantification of fungi in dust. Journal of Environmental Monitoring, 7, 311–318.
Livak, K. J., Flood, S. J., Marmaro, J., Giusti, W., & Deetz, K. (1995). Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods and Applications, 4, 357–362.
Maher, N., Dillon, H. K., Vermund, S. H., & Unnasch, T. R. (2001). Magnetic bead capture eliminates PCR inhibitors in samples collected from the airborne environment, permitting detection of Pneumocystis carinii DNA. Applied and Environmental Microbiology, 67, 449–452.
McDevitt, J. J., Lees, P. S., Merz, W. G., & Schwab, K. J. (2004). Development of a method to detect and quantify Aspergillus fumigatus conidia by quantitative PCR for environmental air samples. Mycopathologia, 158, 325–335.
McDevitt, J. J., Lees, P. S., Merz, W. G., & Schwab, K. J. (2005). Use of green fluorescent protein-expressing Aspergillus fumigatus conidia to validate quantitative PCR analysis of air samples collected on filters. Journal of Occupational and Environmental Hygiene, 2, 633–640.
Olive, D. M. (1989). Detection of enterotoxigenic Escherichia coli after polymerase chain reaction amplification with a thermostable DNA polymerase. Journal of Clinical Microbiology, 27, 261–265.
Roe, J. D., Haugland, R. A., Vesper, S. J., & Wymer, L. J. (2001). Quantification of Stachybotrys chartarum conidia in indoor dust using real time, fluorescent probe-based detection of PCR products. Journal of Exposure Analysis and Environmental Epidemiology, 11, 12–20.
Sachadyn, P., & Kur, J. (1998). The construction and use of a PCR internal control. Molecular and Cellular Probes, 12, 259–262.
Schwab, K. J., Neill, F. H., Le Guyader, F., Estes, M. K., & Atmar, R. L. (2001). Development of a reverse transcription-PCR-DNA enzyme immunoassay for detection of “Norwalk-like” viruses and hepatitis A virus in stool and shellfish. Applied and Environmental Microbiology, 67, 742–749.
Spiess, B., Buchheidt, D., Baust, C., Skladny, H., Seifarth, W., Zeilfelder, U., Leib-Mosch, C., Morz, H., & Hehlmann, R. (2003). Development of a LightCycler PCR assay for detection and quantification of Aspergillus fumigatus DNA in clinical samples from neutropenic patients. Journal of Clinical Microbiology, 41, 1811–1818.
Wasylnka, J. A., & Moore, M. M. (2002). Uptake of Aspergillus fumigatus Conidia by phagocytic and nonphagocytic cells in vitro: quantitation using strains expressing green fluorescent protein. Infection and Immunity, 70, 3156–3163.
Wilson, I. G. (1997). Inhibition and facilitation of nucleic acid amplification. Applied and Environmental Microbiology, 63, 3741–3751.
Zhou, G., Whong, W. Z., Ong, T., & Chen, B. (2000). Development of a fungus-specific PCR assay for detecting low-level fungi in an indoor environment. Molecular and Cellular Probes, 14, 339–348.
Acknowledgements
We thank Brian Schofield, The Johns Hopkins University Bloomberg School of Public Health for assistance with the microscopy. This research was supported in part by a Johns Hopkins Center in Urban Environmental Health pilot grant (ES03818) and a Johns Hopkins NIOSH Education and Research Center Pilot Project Research Training Award (T42/CCT31049-09).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
McDevitt, J.J., Lees, P.S.J., Merz, W.G. et al. Inhibition of quantitative PCR analysis of fungal conidia associated with indoor air particulate matter. Aerobiologia 23, 35–45 (2007). https://doi.org/10.1007/s10453-006-9047-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10453-006-9047-6