
Abstract
We demonstrated the use of multispectral cryo-imaging and software to analyze human mesenchymal stromal cells (hMSCs) biodistribution in mouse models of graft-versus-host-disease (GVHD) following allogeneic bone marrow transplantation (BMT). We injected quantum dot labeled MSCs via tail vein to mice receiving BMT and analyzed hMSC biodistribution in major organs (e.g. lung, liver, spleen, kidneys and bone marrow). We compared the biodistribution of hMSCs in mice following allogeneic BMT recipients (with GVHD) to the biodistribution following syngeneic BMT (without GVHD). Cryo-imaging system revealed cellular biodistribution and redistribution patterns in the animal model. We initially found clusters of cells in the lung that eventually dissociated to single cells and redistributed to other organs within 72 h. The in vivo half-life of the exogenous MSCs was about 21 h. We found that the biodistribution of stromal cells was not related to blood flow, rather cells preferentially homed to specific organs. In conclusion, cryo-imaging was suitable for analyzing the cellular biodistribution. It could provide capabilities of visualizing cells anywhere in the mouse model with single cell sensitivity. By characterizing the biodistribution and anatomical specificity of a therapeutic cellular product, we believe that cryo-imaging can play an important role in the advancement of stem and stromal cell therapies and regenerative medicine.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
There are many published preclinical and clinical studies of stem cell therapies across several different diseases and conditions, including graft-versus-host disease (GVHD), autoimmune diseases, diabetes mellitus, multiple sclerosis, cardiac ischemia, osteoarthritis, cancers, and others.5,8 In this context, it has been recognized that stromal stem cells, such as mesenchymal stromal cells (MSCs) and multipotent adult progenitor cells (MAPCs), have beneficial properties in both trophic and paracrine activities. There is preclinical and clinical evidence that exogenously administered stromal cells can produce and release large amounts of local growth factors and cytokines. In this regard, stromal stem cells can home to sites of injury, such as heart tissue with myocardial infarction,23 traumatic wounds,14 and the central nervous system during stroke,7 where they help establish a regenerative microenvironment conducive to improved healing. In addition, stromal cells can migrate to secondary lymphoid organs (SLOs) including the lymph nodes and spleen and exert potent immuno-regulatory effects.2 Oftentimes, sub-optimal strategies may be used in the clinic because of a lack of preclinical data to guide rational protocol design. There is a significant need to establish functional relationships between infused cells and inflamed tissues, the resulting biodistribution profiles, and the extent and duration of functional benefit of cell products. Indeed, unknown mechanisms of action and lack of a priori information regarding specific homing sites of the administered stem cells represent consistent, critical knowledge gaps in the field of cellular regenerative medicine.1 It is now widely believed that the presence and duration of cell localization to specific sites is a more relevant measure of functional benefit than the administered cell dose. Many cell therapy studies could therefore benefit from a better understanding of the 3D biodistribution of cells following stem cell delivery, subsequent homing to target organs, and potential engraftment.
Typically, standard assays for assessing stem cell homing efficiency utilize one of the following techniques: (1) measuring radioactivity in excised tissues and organs, (2) counting the number of fluorescently labeled cells in the samples using flow cytometry or FACS, (3) measuring the amount of specific DNAs in the samples using PCR or similar assays, and (4) averaging the number of fluorescently labeled cells present in a fixed number of microscopic fields per histological tissue sample.4 However, none of these techniques provide 3D anatomical mapping over a whole animal. This can only be made possible through imaging.
Cryo-imaging system13,22 consists of a fully automated system for repeated physical sectioning and tiled microscope imaging of a tissue block face, providing anatomical brightfield and molecular fluorescence, 3D microscopic imaging with single cell resolution (~ 5–20 μm) and sensitivity over an entire mouse. This makes cryo-imaging advantageous compared to other conventional in vivo imaging modalities such as MRI, PET, SPECT, BLI, FMT (Fluorescent Molecular Tomography), and intravital imaging. Since cryo-imaging uses fluorescence, there are multiple means for labeling cells. Cells can be preloaded with bioconjugated nanocrystal dyes (e.g. quantum dots QDs)), lipophilic dyes (e.g. DiI), and cytoplasmic dyes (e.g. CFDA-SE). The techniques have been widely used for short-term imaging. For long term studies, cells can be bioengineered with reporter gene which generates fluorescent proteins (e.g. EGFP). With dual or triple reporters, one can image the time course of a labeled cell distribution via a method such as BLI or FMT and follow this up with cryo-imaging to exquisitely and accurately localize all cells. Cryo-imaging has been used to investigate stem cell biology and regenerative medicine applications,22,25 phenotyping by spatial mapping of fluorescent reporter gene expression in small animals and embryos,17,21 tumor and metastasis characterization,18etc. Our work here is focused on stem cell applications.
In this paper, we demonstrate application of cryo-imaging for stromal cell imaging in a whole mouse. We focus our analyses on the biodistribution of human MSCs in an established mouse model of GVHD. The analyses include time series of MSC biodistribution in major organs (e.g. lung, liver, spleen, kidneys and bone marrow), and comparison of number of MSCs between allogeneic BMT (with GVHD) and syngeneic BMT (without GVHD) experimental groups.
Materials and Methods
Graft-Versus-Host Disease Mouse Model
We used GVHD as a representative disease model. To induce GVHD in mice, allogeneic BMT (alloBMT), using parent-to-F1 model previously described2 was performed. Briefly, female C57BL/6 J (B6) and B6D2F1 (F1) mice aged 8 to 12 weeks were purchased from Jackson Laboratory (Bar Harbor, ME). Prior to BMT, lethal total body irradiation (14 Gy) was given as a split dose to all F1 recipients. As previously described,2 bone marrow (5 million, 5 M) and T-cells (2 M) collected from either allogeneic (allo) B6 or syngeneic (syn) B6D2F1 donors were suspended in 200 μl Leibovitz L-15 media and injected intravenously into recipient mice on day 0 (D0). T-cell purification was performed by magnetic-bead separation using MicroBeads and the autoMACS system (Miltenyi Biotec, Auburn, CA) with more than 85% of cells obtained being positive for CD4 or CD8 surface antigens.
Stromal Cell Preparation and Administration
For the GVHD model, we used mesenchymal stromal cells (MSCs) as a strategy to prevent GVHD (methods are described in previous publications).2,3,9 Briefly, 1 × 106 human MSCs were labeled with red quantum dots (QD, Qtracker® 625, Life Technologies). The cells were then injected into both allo-BMT (with GVHD) and syn-BMT mice (without GVHD) on Day + 1 or 24 h after BMT. As controls, 1 × 106 unlabeled MSCs were injected into allo and syn BMT recipients. We allowed sufficient time (24 h post injection) for the stromal cells to circulate and home naturally. Animal sacrifice (described in the next section) was performed at different time points following stem cell infusion (24, 48, 72 h). To prepare for cryo-imaging, whole animals were embedded in Optimal Cutting Temperature embedding medium or OCT (Tissue-Tek, Sakura Fintek USA Inc.) inside a custom freezing apparatus, snap frozen in liquid nitrogen, and mounted on to the CryoViz™ specimen stage.
In this study, we used a total of 6 syn mice and 18 allo mice. We infused MSC 24 h after the BMT procedure. After 24 h (T = 0), 2 syn mice (injected with labeled MSCs), 4 allo mice (injected with labeled MSC), and 2 control mice (with unlabeled MSCs) were sacrificed and snap frozen for cryo-imaging. We repeated this procedure for T = 24, 48, 72 h timepoints.
Ethical Statements on Animal Use
Animal work was conducted in the AAALAC accredited Animal Resource Center (ARC) of the Case School of Medicine. The animal health program for all Case owned laboratory animals is directed by the Case ARC Director, W. John Durfee, and was provided by two full-time veterinarians. Mice were used in well-described and reproducible models of murine allogeneic bone marrow transplantation (BMT). All animal experiments received approval from the Institutional Animal Care and Use Committee at Case Western Reserve University IACUC protocol 2010-0076. Detailed descriptions for murine allogeneic BMT can be found in the methods.
Laboratory Animal Euthanasia Guidelines
Following initial sedation with inhaled isoflurane, mice were asphyxiated by a lethal overdose of CO2 followed by bilateral pneumothoracies or terminal eye bleed. To minimize damage to tissue, euthanasia by CO2 was best procedure available. The use of isoflurane sedates the animal before the administration of CO2. The guidelines were approved by Case Western Reserve University IACUC protocol 2010-0076.
Human Mesenchymal Stromal Cell Source
Human MSCs were derived from BM aspirates from healthy donors.3 Patients were consented for the procedure in accordance with the Institutional Review Board of University Hospitals Case Medical Center (UHCMC IRB protocol 09-90-195). Specimens were collected and processed by the Hematopoietic Stem Cell Facility of the Case Comprehensive Cancer Center. Adult volunteer donors underwent BM aspiration (10–30 ml) under local anesthesia. Following cell culture processes, human MSC phenotype was confirmed by morphology, flow cytometry (CD45–CD105 + CD90 + CD80–CD73 + HLA-I+), and in vitro differentiation into osteoblasts, chondroblasts and adipocytes.9
Multipotent Adult Progenitor Cell Biodistribution Experiment
We repeated the experiment using human Multipotent Adult Progenitor Cells (MAPCs). MAPCs are stromal cells that possess tissue regenerative and immunomodulatory properties similar to MSCs.1 Unlike MSCs, MAPCs have broader differentiative potentials into cellular elements of all primitive germline layers including mesoderm, endoderm, and ectoderm. For this experiment, we injected approximately half a million red QD labeled MAPCs to allo mice (n = 4). We also injected another half a million MAPCs that were labeled with SPECT tracers (Tc-99m HMPAO) to the mouse. Since we aimed to compare biodistribution of fluorescently labeled MAPCs and MSCs in the disease model, SPECT results were excluded from this report. Athersys Inc. (Cleveland, OH) generously provided us MultiStem™, a commercialized MAPC-based product, to study the stem cell biodistribution.
Cryo-imaging
Cryo-imaging (CryoViz™, BioInVision, Inc.)13,21,22,29 was used to acquire image data for our experiments. The CryoViz™ cryo-imaging system consists of a digital cryo-microtome housed within a − 20 °C freezer chamber, a microscopic imaging system consisting of a high NA objective, a low-noise, cooled camera, a robotic XYZ positioner, and Programmable Logic Controller (PLC)-based control. CryoViz™ can section accurately between 2 and 200 µm (Z) and create tiled block face images all the way down to ~ 2 µm in-plane (XY) pixel size. Fluorescence images for this study were acquired using a filter cube with 460–490 nm excitation, dichroic, and > 510 nm long-pass green fluorescent protein (long-pass GFP) emission filter followed by voltage controlled electronic RGB LCD filter that allows for capture of the R, G, and B channels separately to form an RGB color image. Brightfield color images were acquired with white light illumination followed by the RGB LCD filter described above. For whole mouse imaging, we sectioned at 40 µm with 10.5 µm pixels in-plane, enabling single cell detection sensitivity.
Image Analysis and Visualization Software
We developed an interactive user interface to provide qualitative and quantitative information from stem cell imaging volumes. Our preprocessing and stem cell detection algorithm is described in detail elsewhere.22,29 The algorithm was designed to automatically search for red fluorescently labeled cells in the cryo-imaging data. In addition to “cell count” obtained from algorithm, we also used “integrated intensity” to assess the stem cell biodistribution. To obtain integrated intensity, we applied the top-hat transformation to the red channel of the fluorescent images. This process removes the autofluorescence signal from the red QD signals. Then we applied morphological reconstruction by using the top-hat image as the mask. We used the stem cell image, created by the stem cell detection algorithm, as the marker. On this transformed image, we applied thresholding to remove low intensity pixels that are likely to be noise. Finally, we summed up all remaining intensities of pixels belonging to the same cell. The number represents integrated intensity for a cell or a cell cluster. The stem cell detection and analysis software were implemented using Matlab platform (The MathWorks Inc., MA). Example visualizations are given in figures and supplemental materials. All 3D visualizations were rendered using Amira platform (Thermo Fisher Scientific).
Human analysts employed semi-automatic segmentation tools to label organs of interest from the whole mouse data. Since tibia and femur bone marrow accounts for 11.9% of total body bone marrow content,24 we used this assumption to estimate cell detection in total body bone marrow. In our study, qualitative assessments for stem cell presence in brain, spinal cord, muscle, heart, adipose tissues, reproductive tract and GI-tract were also examined. Number of false positives were estimated using cell counts in the control group (mice in control group were injected with unlabeled cells). Statistical testing was performed using two-tailed Student’s t test.
Results
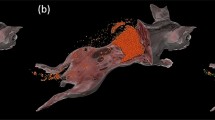
With whole mouse imaging, we can visualize and quantify the distribution of labeled cells throughout a mouse (Figs. 1 and 2, and Supplemental Videos 1–3). Stromal stem cells were found in most of the tissues, but most commonly in lung, liver, spleen, kidneys, and bone marrow, all preferential sites for stem cell homing.12 Following the injection of 1 × 106 MSCs and performing cryo-imaging 24 h later, we found on average 305,515 cells ± 31,938 standard error (SE) throughout the whole mouse (n = 4, 1 syn + 3 allo). The number of detected cells were: liver 153,999 ± 18,559 cells, lung 91,033 ± 29,667 cells, bone marrow 4482 ± 1268 cells, spleen 3130 ± 993 cells, and kidneys 2463 ± 636 cells (mean ± SE). The recovery rate (number of cells found/number cells delivered) was 31% at the 24-h timepoint. We repeated the experiment using human MAPCs (MultiStem™, Athersys Inc) and found the recovery rate to be 58% at the same time point (Fig. 2 and Suppl. Fig. 1). Both types of stromal stem cells were found to home to the bone marrow especially the endosteal region (Supplemental Video 3). These results were not very surprising, because the stromal cells used in this experiment are derived from the bone marrow. There was one mouse that showed a sign of broken spine, surprisingly, we observed a significant number of MAPCs infiltrated the paraspinous muscles near the injury site (Arrow in Fig. 1b and Supplemental Video 2). Moreover, we observed that in mice developing GVHD, stem cell density in brain, spinal cord, heart, adipose tissue and muscle was very low. Specifically, exogenous stem cells in the brain region are mostly found in the surrounding bones such as cranial bone marrow with none found in the brain tissue as such (Supplemental Video 4). Raw data of the brightfield and fluorescent images as well as the stem cell signals in various tissues were added to Suppl. Fig. 2.

Cryo-imaging enables analysis of stromal cell biodistribution anywhere in an entire mouse with single cell sensitivity. In this experiment, red quantum dot labeled mesenchymal stromal cells (MSCs, (a)) and multipotent adult progenitor cells (MAPCs, (b)) were intravenously infused into a GVHD mouse, following which cryo-imaging was performed. Using our specialized software, we could visualize stromal cell biodistribution (yellow beads) anywhere in an entire mouse. Stromal cells were substantially found in lung, liver and spleen. There was one mouse that showed a sign of broken spine, surprisingly, we observed a significant number of MAPCs infiltrated the paraspinous muscles near the injury site (Arrow in (b)). Red quantum dot signal detection algorithm was proposed in the previous work. (Only figure (b), © 2016 IEEE. Reprinted, with permission, from “Automatic Stem Cell Detection in Microscopic Whole Mouse Cryo-Imaging” by P. Wuttisarnwattana et al., in IEEE TMI,29).

Number of cells detected in major organs of GVHD mice. Fluorescently labeled MSCs (1 × 106 labeled cells) and MAPCs (0.5 × 106 labeled cells) were intravenously injected into GVHD mice. At 24 h post injection, cryo-imaging was performed, and number of cells detected in each organ were quantified. The recovery rates (number of cells found/number cells delivered) were 31% for MSCs (1 × 106 labeled cells injected) and 58% for MAPCs (0.5 × 106 labeled cells injected). Note that numbers represent mean ± SE and the number of mice in each group was 4 (n = 4).
The total number of detected human MSCs in vivo decreased over time (Fig. 3). At time points 24, 48, and 72 h post infusion, the average global cell detections were 305,515, 127,106, and 71,203 respectively. We performed exponential fitting to estimate MSC half-life in vivo, which was determined to be 21 h (2 syn + 4 allo for 3 timepoints, n = 18 totally). The average number of false positive detections in control mice implanted with unlabeled fluorescent cells was 6742 (n = 6, across all time points). As compared to the number of detections BMT recipient mice, the number of false positives in a control mouse was negligible (p < 0.05).

Total number of detected human MSCs in the mouse decreased over time. Result from the exponential fitting suggests that the in vivo half-life of hMSCs was ≈ 21 h. We speculate that cells undergo apoptosis and clearance. The number of detections in control mice was significantly low (n = 6, across all time points). Each time point consists of 2 syn + 4 allo (n = 18 totally). (Bar = mean + SE, *p < 0.05).
Volume rendering of detected red fluorescently labeled cells shows that the cell density in lung was much higher than that in other tissues such as the liver and the spleen (Suppl. Fig. 3). Cell density was defined as number of cell detection inside a particular organ (cells) divided by volume of the organ (mm3). The calculated cell densities (cells/mm3) in the organs of interest were consistent with the visualization (Fig. 4).

Cell density in the tissues of interest. Data show that cell density in lung, liver and spleen were significantly higher than those from other tissues of interest. The data suggest that lung, liver, and spleen could be the preferential homing organs in our mouse model. Very high cell density in lung may also suggest pulmonary emboli due to intravenous injection. As compared to cell detection in mice from the experimental group, false positive detection in the control group was very low. Note that the y-axis is shown in log scale.
Another method to assess cell density for a particular organ is via the integrated intensity which is closely correlated with the fluorescent activity. To calculate, an average integrated intensity, we firstly calculated the summation of the integrated intensity of all detected cells inside the organ and then divided by number of cells. The algorithm to find an integrated intensity was described in the Image analysis sub-section. Interestingly, the average integrated intensity per detection in the lung was 233, about three times higher than values measured in the liver, spleen, bone marrow, and kidneys of 78, 87, 55, and 86 respectively (Fig. 5). We also examined total fluorescent activity using total integrated intensity inside an organ. Among five organs of interest (lung, liver, spleen, kidneys and bone marrow), 97% of total integrated intensity of the detected cells were in the lung and 95% of total cell detections were in the liver (Suppl. Fig. 4).

Integrated cell signal per detection shows a much higher intensity in the lungs where cells were clumped than in the other organs where there were single cells. In all organs except the lung, integrated intensity per detection corresponds to the average intensity of isolated cells (≈ 77 grayscale intensity). The results show that single cell intensity in each of the organs (except the lung) was consistent. In lung, the average intensity of detections was much higher as compared to those in other organs (≈ 233 grayscale intensity), corresponding to a factor of 3, suggesting 3 cells per cluster on average in the lung. Bars represent mean ± SE. Data were collapsed across all time points.
Interestingly, we observed redistribution of cells from lung to the liver over time; results show that the integrated intensity in lung decreased over time, whereas that in the liver tended to increase over time (Fig. 6). At 24 h post infusion, the average percentage of total integrated intensity in lung and liver was 71 ± 9% and 18 ± 8%, respectively (mean ± SE). At the 72-h time point, the percentage of integrated intensity in lung and liver changed to 36 ± 10% and 44 ± 10%, respectively.

Cell clusters in the lungs dissociate and migrate mostly to the liver over time. The figure shows change in the integrated intensity in lung and liver over time, as a percentage of total integrated intensity in the five major organs at the respective time point. The percentage of integrated intensity in the lung clearly decreased over time, while the percentage in the liver increased. The result suggests that cluster of cells in the lung could eventually dissociate to single cells and pass through the pulmonary capillary bed. Interestingly, the integrated intensity in the liver increased over time. Each data point represents mean ± SE.
We observed that the total number of MSC detections in allo BMT recipients was greater than that present in syn mice (Fig. 7a). This may suggest that the MSCs became activated and persist longer in vivo under the GVHD environment. Also, we observed that the cell count in the liver of the allo mice tended to be greater than that of the syn mice at the earlier time points (Fig. 7b). This also may suggest that liver could be the target organ for our disease model. Number of detections in both groups was significantly higher than those in the control group (p < 0.05).

Number of cell detections in syn and allo groups. Panels show numbers of cell detections in whole mouse (a) and in livers (b). As compared to the allo group, the number of total cell detection in the syn group (a) was lower at the earlier time point. We hypothesize that small numbers of administered MSCs escape apoptosis and migrate to sites of injury and inflammation and provide beneficial effects in the setting of GVHD. In our experiments, the liver could be damaged by either the effects of total body irradiation or being a GVHD target organ, or both. The activated MSCs then eventually accumulated in liver in response to the injury and provided healing effects. The number of false detections in the control group was negligible (bar = average ± SE).
Discussion
We have demonstrated the utility of cryo-imaging in regenerative cell therapy, taking MSC therapy in GVHD mice as an example. Our methodology has several innovative features. Analyzing stromal cell biodistribution with single cell sensitivity over a mouse-sized field-of-view is possible only through cryo-imaging (Fig. 1 and Supplemental Videos 1, 2). We developed specialized software to detect signal from red fluorescently labeled MSCs. We used an approach which rendered stromal cells as individual beads with a size sufficient to show up in a 3D visualization with a fixed pixel size. We show that cells were not “lost” in the visualization process of zooming out to coarser resolutions. The interactive 3D visualization enables one to zoom in to see individual cells in a particular organ of interest or zoom out for global biodistribution visualization. We encourage readers to examine our videos generated from the cryo-imaging data in the supplemental media.
Preferential homing sites of MSCs were identified. In our GVHD mouse model, lung, liver and spleen had a significantly higher cell density than other tissues in the body (Fig. 4 and Suppl. Fig. 3). Additionally, cell intensity in lung was significantly higher than that for other tissues (liver, spleen, kidneys and bone marrow) (Fig. 5) at early time points consistent with the lung representing the first microvascular bed encountered by cells injected via the tail vein of a mouse.10 The integrated intensity of cell detections in lung was about three times higher than that in other tissues. This can suggest that one cell cluster (one detection) in lung consists of three cells. While the initial lung signal was represented by clusters of cells, we found that these clusters could later dissociate into single cells prior to redistribution to other organs. The majority of exogenous MSCs were found in the lung and the liver (Suppl. Fig. 4) in which the integrated intensity and number of detections accounted for about 95% among the organs of interest. In the case of the lung where cell clusters were found, we deem integrated intensity to be more appropriate than cell number count because it directly relates to fluorescent activity of the cells.
A greater number of cells was detected in whole mice from the allo group compared to mice from the syn control group at the early time point (Fig. 7). We hypothesize that this is due to activation of the MSCs in the allo group due to the development of GVHD, which gives rise to a protective effect in the MSC population. In this context, small significant number of administrated MSCs escape apoptosis and migrate to sites of injury and inflammation and provide beneficial effects in the setting of GVHD. For example, we have previously shown that MSCs also migrate to the splenic marginal zone after allo-BMT where they exert potent anti-proliferative effects and modulate the expansion of alloreactive, donor-derived T cells in the white pulp.2 Our observation is consistent with those in the literature showing that activated cells have a longer live span in vivo.28
Our results suggest that MSCs were activated and migrated to injured liver. We found that MSCs were found substantially in liver. Also, there was a relatively higher number in the allo group as compared to in the syn group. In our experiments, liver could be damaged by either the effects of total body irradiation or being a GVHD target organ, or both. We speculate that with the systemic inflammation, the exogenous MSCs could sense sites of inflammation and shift to an anti-inflammatory state with healing potential. The activated MSCs then eventually accumulated in liver in response to the injury. Our previous study revealed that liver was damaged in the GVHD mouse model and the MSC treatment was able to reverse the symptom.2 The results were consistent with other studies and the molecular mechanism was described elsewhere.16,28
Interestingly, we observed that stem cell densities in brain, spinal cord, kidney, heart, adipose tissue and muscle were very low, regardless of high cardiac output to these tissues. We typically found only ~ 100 cells (out of one million injected) in the brain tissue, indicating that the blood brain barrier was relatively impervious to the infusion of MSCs in our experiments. Supplemental Video 4 shows that MSCs were located in the bone marrow of the skull rather than in the brain tissue. Low counts were also found in the kidneys, another highly perfuse organ. We conclude that the stem cell biodistribution is not simply related to blood flow. Instead, cells preferentially deposit in specific organs likely driven in part by inflammation or immune dysregulation.
Moreover, we surprisingly found that the stromal cells (MAPCs) infiltrated into the paraspinous muscles of a mouse inadvertently injured during immobilization required for intravenous injection (Fig. 1b and Supplemental Video 5). Healthy muscle is expected to exhibit much lower stem cell density (unpublished work). This supports the hypothesis that stem cells can sense injury and specifically home to the site of injury where they aid the healing process.20
Cryo-imaging compares favorably to other small animal imaging techniques. Conventional in vivo imaging modalities such as MRI, PET, SPECT, BLI (Bioluminescence imaging), and intravital imaging are useful for stem cell studies, however these methods have limitations. With loading of super-para-magnetic iron nanoparticles, one can detect an MRI susceptibility artifact arising from a single cell in homogenous brain tissue,6 but detection in other tissues and quantification are problematic. Using a mouse model of demyelinization, a report from Bulte’s group27 shows that super-paramagnetic iron oxide signal did not reflect the distribution of lacZ-labeled neuronal stem cells at 6 days, due to signal loss with proliferation and differentiation. When cells die, macrophages could engulf dead cells containing the magnetic particles, which could lead to misleading interpretation of cell homing. PET and SPECT have been used to image stem cells in vivo, but resolution, sensitivity, and cell quantification are limited. Pre-loading cells with Technetium-99m (Tc-99m) enables sensitive detection over the entire animal, but the signal is only detectable for 2–3 days. Reporter gene approaches such as BLI have good sensitivity, with detection of 10 cells at the skin surface,19 however resolution is limited and interior cells are far less detectable. Intravital imaging gives single cell imaging within live hosts, but over very small volumes.15 None of these methods are suitable for detecting and quantifying, say, less than a 1000 cells within a deep organ such as the spleen. In general, small animal imaging techniques (MRI, PET, SPECT, BLI, etc.) do not provide micron-scale resolution, color contrasts, sensitive imaging of weak fluorescence, and single cell detection. Cryo-imaging alone provides 3D microscopic-resolution, anatomical color and molecular fluorescence volumes with single fluorescent cell sensitivity over fields-of-view as large as an entire mouse or a rat. In other words, the combined features of high resolution, large volume of view, and single cell sensitivity of cryo-imaging make it an ideal imaging modality for detection and analysis of stem cell biodistribution.
Quantum dots (QDs) are excellent fluorescent labeling probes for analyzing short-term biodistribution of stem cells using the cryo-imaging system. They give continuously bright fluorescent signals, resist photo-bleaching, and have large absorption coefficients across a broad spectral excitation range. They appear to be non-toxic and exert minimal impact on stem cell functions.20 It has been reported that QDs are not leaked out of intact cells and taken up by adjacent cells.20 but here is a potential that a dead labeled cells could be engulfed by a macrophage, leaving a false bright signal. QD labeling is for short term imaging. In our experiment, we found that fluorescence of the probes could be seen up to a week after delivery. For longer term studies of engraftment, we recommend the use of reporter gene technique where cells can stably create fluorescent proteins in vivo for weeks and even months.
Factors which might be construed to affect cell counts, do not significantly affect results. First, if there is a varying amount of QD cell labeling, it could affect detection. However, we strictly followed the cell labeling protocol suggested by the QD manufacturer (QTracker®, Thermo Fisher Scientific). In our experiment, good labeling efficacy and homogeneity of intensity was confirmed by flow cytometry result (data not shown). Second, as tissues have different optical properties, cell detection efficacy might vary. However, we observed that signal of a single cell could be seen in all tissue types regardless of the differences in tissue optical properties. We examined raw cryo-imaging data and virtually found cells in all tissue types. This was true even in the highly attenuated tissues such as bone marrow, fatty tissues, and heart. We believe that this was due to the fact that cell brightness was sufficiently high before the injection. Third, as subsurface cells can be seen, this might affect cell counts. However, in our software, connected fluorescence spots in 3D volume are counted as a single cell cluster,26 eliminating this potential confound. Fourth, photo-bleaching could affect detection. However, QDs are known to be resistant to photo-bleaching,11,20 and none was observed in our experiments. Moreover, we have never observed photobleaching in cryo-imaging, likely because the sample is frozen and because fluorophores are protected from exposures by intervening tissues prior to sectioning. In one surprising experiment, every few seconds, we imaged a frozen tissue block face containing GFP-labeled cancer cells for an hour without sectioning and saw no diminution of signal. Previously, we have improved stem cell detection algorithm using alpha multiplier technique to account for signal variation of the stem cells.29 The program could robustly detect low intensity signals (1/3 of the standard brightness) with 99% accuracy. Also, please note that in spite of these potential factors, in a heroic experiment, we were able to detect 94% of the 100,000 stem cells implanted in a lung, a percentage within experimental accuracy (manuscript in prep).
In conclusion, we have demonstrated applications of cryo-imaging in whole body and local (i.e. organ and tissue) stem cell biodistribution, and dynamics of stem cell homing patterns. We believe that the cryo-imaging technology will have a great impact on the advancement of stem cell therapy and regenerative medicine.
References
Auletta, J. J., K. R. Cooke, L. A. Solchaga, R. J. Deans, and W. van’t Hof. Regenerative stromal cell therapy in allogeneic hematopoietic stem cell transplantation: current impact and future directions. Biol. Blood Marrow Transplant. 16:891–906, 2010.
Auletta, J. J., S. K. Eid, P. Wuttisarnwattana, I. Silva, L. Metheny, M. D. Keller, R. Guardia-Wolff, C. Liu, F. Wang, T. Bowen, Z. Lee, L. A. Solchaga, S. Ganguly, M. Tyler, D. L. Wilson, and K. R. Cooke. Human mesenchymal stromal cells attenuate graft-versus-host disease and maintain graft-versus-leukemia activity following experimental allogeneic bone marrow transplantation. Stem Cells 33:601–614, 2015.
Auletta, J. J., E. A. Zale, J. F. Welter, and L. A. Solchaga. Fibroblast growth factor-2 enhances expansion of human bone marrow-derived mesenchymal stromal cells without diminishing their immunosuppressive potential. Stem Cells Int. 2011:235176, 2011.
Barbash, I. M., P. Chouraqui, J. Baron, M. S. Feinberg, S. Etzion, A. Tessone, L. Miller, E. Guetta, D. Zipori, L. H. Kedes, R. A. Kloner, and J. Leor. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation 108:863–868, 2003.
Barry, F. P., and J. M. Murphy. Mesenchymal stem cells: clinical applications and biological characterization. Int. J. Biochem. Cell Biol. 36:568–584, 2004.
Bulte, J. W., I. D. Duncan, and J. A. Frank. In vivo magnetic resonance tracking of magnetically labeled cells after transplantation. J. Cereb. Blood Flow Metab. 22:899–907, 2002.
Chen, J., Y. Li, L. Wang, Z. Zhang, D. Lu, M. Lu, and M. Chopp. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 32:1005–1011, 2001.
Copelan, E. A. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 354:1813–1826, 2006.
Dominici, M., K. Le Blanc, I. Mueller, I. Slaper-Cortenbach, F. Marini, D. Krause, R. Deans, A. Keating, D. Prockop, and E. Horwitz. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317, 2006.
Fischer, U. M., M. T. Harting, F. Jimenez, W. O. Monzon-Posadas, H. S. Xue, S. I. Savitz, G. A. Laine, and C. S. Cox. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 18:683–691, 2009.
Gao, X., L. W. Chung, and S. Nie. Quantum dots for in vivo molecular and cellular imaging. Methods Mol. Biol. 374:135–145, 2007.
Gao, J., J. E. Dennis, R. F. Muzic, M. Lundberg, and A. I. Caplan. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs 169:12–20, 2001.
Gargesha, M., M. Q. Qutaish, D. Roy, G. J. Steyer, M. Watanabe, and D. L. Wilson. Visualization of color anatomy and molecular fluorescence in whole-mouse cryo-imaging. Comput. Med. Imaging Graph. 35:195–205, 2011.
Li, H., X. Fu, Y. Ouyang, C. Cai, J. Wang, and T. Sun. Adult bone-marrow-derived mesenchymal stem cells contribute to wound healing of skin appendages. Cell Tissue Res. 326:725–736, 2006.
Myers, J. T., D. S. Barkauskas, and A. Y. Huang. Dynamic imaging of marrow-resident granulocytes interacting with human mesenchymal stem cells upon systemic lipopolysaccharide challenge. Stem Cells Int. 2013:656839, 2013.
Najar, M., M. Krayem, M. Merimi, A. Burny, N. Meuleman, D. Bron, G. Raicevic, and L. Lagneaux. Insights into inflammatory priming of mesenchymal stromal cells: functional biological impacts. Inflamm. Res. 67:467–477, 2018.
Powell, K. A., and D. Wilson. 3-dimensional imaging modalities for phenotyping genetically engineered mice. Vet. Pathol. 49:106–115, 2012.
Qutaish, M. Q., K. E. Sullivant, S. M. Burden-Gulley, H. Lu, D. Roy, J. Wang, J. P. Basilion, S. M. Brady-Kalnay, and D. L. Wilson. Cryo-image analysis of tumor cell migration, invasion, and dispersal in a mouse xenograft model of human glioblastoma multiforme. Mol. Imaging Biol. 14:572–583, 2012.
Rabinovich, B. A., Y. Ye, T. Etto, J. Q. Chen, H. I. Levitsky, W. W. Overwijk, L. J. N. Cooper, J. Gelovani, and P. Hwu. Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proc. Natl. Acad. Sci. USA 105:14342–14346, 2008.
Rosen, A. B., D. J. Kelly, A. J. T. Schuldt, J. Lu, I. A. Potapova, S. V. Doronin, K. J. Robichaud, R. B. Robinson, M. R. Rosen, P. R. Brink, G. R. Gaudette, and I. S. Cohen. Finding fluorescent needles in the cardiac haystack: tracking human mesenchymal stem cells labeled with quantum dots for quantitative in vivo three-dimensional fluorescence analysis. Stem Cells 25:2128–2138, 2007.
Roy, D., M. Gargesha, G. J. Steyer, P. Hakimi, R. W. Hanson, and D. L. Wilson. Multi-scale characterization of the PEPCK-C mouse through 3D cryo-imaging. Int. J. Biomed. Imaging 2010:105984, 2010.
Roy, D., G. J. Steyer, M. Gargesha, M. E. Stone, and D. L. Wilson. 3D cryo-imaging: a very high-resolution view of the whole mouse. Anat. Rec. 292:342–351, 2009.
Schenk, S., N. Mal, A. Finan, M. Zhang, M. Kiedrowski, Z. Popovic, P. M. McCarthy, and M. S. Penn. Monocyte chemotactic protein-3 is a myocardial mesenchymal stem cell homing factor. Stem Cells 25:245–251, 2007.
Shaposhnikov, V. L. Distribution of bone-marrow cells in the mouse skeleton. Bull. Exp. Biol. Med. 87:510–512, 1979.
Steyer, G. J., F. Dong, L. Kanodia, D. Roy, M. Penn, and D. L. Wilson. Detection and quantification of fluorescent cell clusters in cryo-imaging. Int. J. Biomed. Imaging 2012:698413, 2012.
Steyer, G. J., D. Roy, O. Salvado, M. E. Stone, and D. L. Wilson. Removal of out-of-plane fluorescence for single cell visualization and quantification in cryo-imaging. Ann. Biomed. Eng. 37:1613–1628, 2009.
Walczak, P., D. A. Kedziorek, A. A. Gilad, B. P. Barnett, and J. W. Bulte. Applicability and limitations of MR tracking of neural stem cells with asymmetric cell division and rapid turnover: the case of the shiverer dysmyelinated mouse brain. Magn. Reson. Med. 58:261–269, 2007.
Weiss, A. R. R., and M. H. Dahlke. Immunomodulation by mesenchymal stem cells (MSCs): mechanisms of action of living, apoptotic, and dead MSCs. Front. Immunol. 10:1191, 2019.
Wuttisarnwattana, P., M. Gargesha, W. van’t Hof, K. R. Cooke, and D. L. Wilson. Automatic stem cell detection in microscopic whole mouse cryo-imaging. IEEE Trans. Med. Imaging 35:819–829, 2016.
Acknowledgments
The research was supported by an Ohio Third Frontier Wright Projects Program award (OTFWPP) (D.L.W., co-I), National Center of Regenerative Medicine Pilot Grant (K.R.C.), the Ohio Board of Regents (K.R.C.), the Meredith Cowden Foundation (K.R.C.), The Thailand Research Fund MRG6080218 (P.W.), and the National Institute of Health through R42-CA124270, R41HD063241-01 (D.L.W.).
Conflict of interest
Dr. Gargesha (M.G.) declares that he is an employee of BioInVision Inc. which is a company that manufactures CryoViz™, a system that utilizes the cryo-imaging principle as the core technology. The rest of the authors (P.W., S.E., K.R.C., D.L.W.) declare that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Smadar Cohen oversaw the review of this article.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wuttisarnwattana, P., Eid, S., Gargesha, M. et al. Cryo-imaging of Stem Cell Biodistribution in Mouse Model of Graft-Versus-Host-Disease. Ann Biomed Eng 48, 1702–1711 (2020). https://doi.org/10.1007/s10439-020-02487-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-020-02487-z