
Abstract
Our hemostatic system, when called to action, depends on the complex arrangement of a tightly regulated and extensive network of molecules with versatile functionality. Experimental methods have demonstrated marked improvement through enhanced condition-control and monitoring. However, this approach continues to provide limited explanations of the role of individual elements or of a specific component within the entire system. To fill this void, multiscale simulations based on high throughput computing and comprehensive mathematical models are showing their strength in not only revealing hidden physiological mechanisms but also predicting pharmacological/phenotypical outcome in hemostasis reactions based on quantitative analysis. In this review article, we present up-to-date computational methods that simulate the process of platelet adhesion and thrombus growth, compare and summarize their advantages and drawbacks, verify their predictive power, and project their future directions. We provide an in-depth summary of one such computational method—Platelet Adhesive Dynamics (PAD)—and discuss its application in simulating platelet aggregation and thrombus development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Platelet adhesion and thrombus formation are critical, self-defensive mechanisms during the hemostatic response to prevent excessive blood loss during body injuries. When an unactivated, flowing platelet encounters an elevated local concentration of soluble platelet activators (e.g., thrombin or ADP, released at the site of vessel injury12), or physically contacts an exposed subendothelial layer through formation of surface receptor–ligand bonds (e.g., GPIbα–vWF–A1 bond36) it initiates translocation on an injured vessel wall, platelet activation, and subsequently, firm adherence to the vessel wall. Further development of a thrombus involves platelet–platelet interaction via GPIbα–fibrinogen/vWF bridging,67 as well as formation of a fibrin network.39 Defects in hemostasis arising from genetic mutations or environmental stress can result in bleeding disorders (failure to stop bleeding) or thrombosis (undesirable blood clotting), with symptoms ranging from mild to life threatening.11 Pathologies of hemostasis affect more than 2% of the US population. Computer simulations of hemostasis have been developed to supplement knowledge gained through experimental and clinical research. For more than a decade, simulation studies have evolved to study platelet aggregation and thrombus formation under increasingly realistic conditions (e.g., complicated vascular geometry, multiple ligand-binding kinetics, and comprehensive coagulation cascade system79). Though model simplification, assumption, and approximation remain unavoidable, researchers are building more accurate models with the aid of enhanced computing power and an improved understanding of the underlying biological processes.
Current multiscale computational methods applied to study these delicate physiological processes utilize different compositions of several fundamental yet important modules, such as fluid mechanics, coagulation cascade, cell mechanics and receptor–ligand binding. Each of these modules focuses on a specific range of the length/time scales (Fig. 1). More detailed description of each module can be seen in Table 1. A reasonable platelet aggregation model usually comprises two or more of these inter-correlated modules to achieve the multiscale model structure, and such inter-correlation is usually achieved by variable transmission. For example, Alber and colleagues have established a 2D thrombus development model,80 which considers both the hemodynamics and coagulation cascade reactions, but is not involved in cell mechanics and receptor–ligand binding. Pivkin et al.59 have developed a 3D model that incorporates fluid mechanics and physical cell particle modules where a platelet-rich thrombus is growing inside a cylindrical vessel, but the adhesion model is based on threshold assumptions that have not been experimentally supported. Fogelson and coworkers17,74 developed a continuum platelet aggregation model by considering only the fluid phase, where the effect of platelet adhesion was simulated by a force distribution generated from elastic linkage between cells onto the fluid (plasma). The model was later modified to incorporate the platelet-wall interaction18 and a partial coagulation cascade.37 Our group has constructed a 3D platelet aggregation model (Platelet Adhesive Dynamics, or PAD) with ellipsoid platelet particle aggregates via receptor–ligand binding under simple shear flow.50,51 However, this model has not yet incorporated the coagulation/platelet activation effect. Validation of these mathematical models is often done by comparing simulation results with in vivo/in vitro experimental data. The experimental observations can cover a wide range of types, including cellular response to signal molecules, visualization of cell motion in microfluidic devices and confocal microscopy of in vivo thrombus development.

The summary of the systematic structure of different methods, modules and models in current studies of multiscale modeling of platelet aggregation and thrombus development. Here, each module utilizes a group of selected methods (usually representative governing equations), and one computational model usually contains several organized modules
Hydrodynamics
Hydrodynamics and blood rheology are critical for maintaining normal hemostatic function. The margination of platelets, dependent on factors such as finite platelet size,72 large volume fraction of red blood cells (RBCs), the relatively high stiffness of platelets (compared to highly deformable RBCs)55,85 and physiological shear stress in the blood flow,84 tends to cause platelets to be concentrated in the peripheral region near the vessel wall. This enables efficient attachment of platelets onto the injured vessel wall which results in reduced bleeding time. The non-Newtonian nature of blood, primarily resulting from fibrinogen in the plasma47 and deformable RBCs,57 may also entrap activated platelets in vortices and recirculation regions to form clots.15 These effects are all consequences of hydrodynamics.
Hydrodynamic modules directly determine the level of realism of the physical model. Though most existing methods utilize the Navier–Stokes Equation as the governing equation, different model settings greatly affect the simulation power to recreate the realistic physiological environment. Commercially available software packages are usually designed based on computational fluid dynamics (CFD) methods, which were introduced in the 1990s1 and are potentially powerful tools to elucidate some of the hemodynamic challenges. Nowadays, CFD has evolved to be combined with image-based,71 personalized65 techniques to study individual symptoms. However, when studying flow-induced blood clotting, an approach based on a single-phase continuum assumption such as that employed by many commercial CFD packages may inaccurately represent the nature of blood flow during hemostasis. It may also fail to model the multiscale nature when characterizing blood constituents (soluble proteins and cells), which change their properties in response to biochemical/mechanical signals. Recent progress on advanced computational methods as well as high-performance computing has aided researchers in developing more versatile approaches such as Stokesian Dynamics Methods (SDM), Dissipative particle dynamics (DPD) and the Completed Double Layer Boundary Integral Equation Method (CDL-BIEM).
The Stokesian Dynamics Method was developed by Brady et al.7 and is capable of simulating the motion of a group of highly concentrated, finely shaped (mostly spheres) solid particles in a low Reynolds number suspension fluid while accurately accounting for the hydrodynamic interaction between them. Yamaguchi’s group developed a thrombus development model based on SDM and successfully confirmed that the development of thrombus formation in height requires not only von Willebrand factor, but also fibrinogen.53 Later, they incorporated the presence of RBCs and argued that RBCs play a role in hemostasis by promoting horizontal spreading of the growing thrombus.54 Another method that is also capable of dealing with a large number of suspension particles, but not limited to the Stokes flow condition, is the Lattice Boltzmann Method (LBM). LBM is related to the Molecular Dynamics (MD) method and has successfully simulated multiphase blood flow with a hematocrit value of 40% and platelets28,46,77 though no effort has been devoted to thrombus formation yet.
The DPD method, on the other hand, simulates both plasma and platelets as virtual discretized particles that move according to Newton’s Law, thus sharing similarities with MD as well. Though discretization of the continuum Navier–Stokes equation is used, the DPD method has been shown to compare well with Navier–Stokes solutions.82 Tsuda and colleagues applied the DPD method to construct the thrombosis development process through platelet accumulation onto the vessel wall.16 Pivkin et al.60 also utilized DPD to study the enhanced diffusivity of platelets caused by RBCs on platelet aggregation.
The Completed Double Layer Boundary Integral Equation Method is a boundary integral method that is used to solve an integral form of Stokes equation under low Reynolds number flow. Kim and Karilla discuss the theory detailing the development of this technique in their text32 and the derivation of this solution method as applied to particulate flow in a half space is fully described by Phan-Thien et al.58 As a boundary integral method, CDL-BIEM is specialized to solve the mobility problem for arbitrarily shaped solid particles represented by mesh-grid structures, for instance, when an ellipsoid platelet is flowing close to the vessel wall. As a result, CDL-BIEM is able to explore the shape factor of a platelet during thrombosis.48 Further, Mody et al.51 conclude that the platelet–platelet hydrodynamic collision patterns are significantly different when implementing platelets in their natural ellipsoid shape rather than a spherical shape.
Coagulation Cascade
The blood coagulation cascade is an essential component of hemostasis reactions. It has two initially independent pathways, namely the contact activation pathway (intrinsic pathway) and the tissue factor pathway (extrinsic pathway).22,41,45 These two pathways merge at the point of activating procoagulant protein factor X into factor Xa, which forms the tenase complex and mediates thrombin generation. Thrombin in turn acts as a serine protease that converts soluble fibrinogen into insoluble strands of fibrin leading to formation of fibrin network. Fibrin, together with platelets, support the mechanical structure of a thrombus.73
Over the past two decades, the systems biology approach has thrived and become a common tool for studying blood coagulation and thrombosis. Ataullakhanov and Panteleev3 classified coagulation models into two sub-categories: (1) the reaction sub-model and (2) the physical sub-model. The reaction sub-model is composed of independent reactions (represented by a list of ODEs and PDEs) that when combined, encompass the coagulation cascade, while the physical sub-model contains the spatial properties that take the mass transport under flow dynamic into consideration. The reaction sub-model has been widely studied by computational biologists.13 For example, tissue factor initiated blood coagulation was explored,42 and the kinetics of thrombin generation was revealed and compared with high throughput experimental results.8 The systems biology approach is also capable of identifying potential “fragile points” inside the cascade which serve as good targets for therapeutic strategies44 despite the remaining parametric and structural uncertainty of the model.43,44 In order to study thrombus development, both sub-models should be incorporated into the coagulation module. Though most researchers using the systems biology approach to study the coagulation cascade don’t consider the physical existence of the developing thrombus, and most researchers studying platelet aggregation/thrombus formation do not take into detailed consideration either thrombin-mediated platelet activation or the fibrin network, there are some groups that attempt to couple those two complex systems together.
One of the first models that incorporated the coagulation cascade (especially the extrinsic pathway in the following cases) and platelet deposition was introduced by Kuharsky et al.37 in 2001. The model contains both plasma-phase and membrane-phase reactions, and accounts for chemical and cellular transport by flow and diffusion.37 It predicts the threshold manner of change of thrombin production due to the increasing number of tissue factor binding sites. It also argues that the inhibition of the activity of TF:VIIa enzyme complex is dominant by physical blockage of platelets onto the subendothelium, rather than chemical inhibitors.37 By using a similar approach, Fogelson et al.21 studied the influence of flow-mediated transport on the initiation and inhibition of coagulation. Later, Leiderman and Fogelson established a spatial–temporal model to study platelet activation and blood coagulation under flow40 and they discovered the dependence of FXI dependent thrombin generation on platelet count.19 The above models treat the different status of platelets (activated, non-activated, subendothelium bound, etc.) as “chemical-like,” which describes the physical existence of platelets by their spatial concentration profile.
On the other hand, Xu et al. implemented the tissue factor pathway described in Jones and Mann30 for the generation of thrombin involving the activation of factors IX, X, V, VIII, VIIIa–IXa, and factors Va–Xa (prothrombinase) into their thrombosis model.78 In 2010, Xu et al.80 extended their coagulation model into both solution-phase and membrane-phase reactions, the total number of factors being considered in the model also increased by several-fold. Their model considers the coagulation process as a source of cell status change78 but no detailed mechanical properties of the fibrin network were considered.
Though all the existing coagulation modules remain within 2D space, they are to be extended into 3D. Further, they will be more commonly applied to future simulation studies of thrombosis, as the importance of the mechanical structure of the fibrin network to the mature thrombus function is being revealed and receiving more attention.20,75
Receptor–Ligand Binding
The initiation of thrombus development starts with tethering of circulating platelets onto the exposed subendothelial layer at the site of a blood vessel injury via bonding between the α subunit of GPIb receptors on the platelet surface and the A1 domain of subendothelial collagen-bound multimeric plasma glycoprotein von Willebrand Factor (vWF).66,67 Such GPIbα–vWF–A1 tethering leads to platelet translocation on the subendothelial surface, which prolongs the duration of the platelet-vessel contact, helping platelets gain a sufficient number of biological/biochemical signals to trigger platelet activation.12,36 This leads to the formation of other receptor–ligand bonds such as integrin α2β1 with subendothelial collagen to support firm adhesion of the platelet31 or αIIbβ3 to another αIIbβ3 molecule on the surface of other platelets via fibrinogen or vWF to promote platelet aggregation.6
Most existing thrombus development models have oversimplified this critical module either using “guessed” values for non-experimentally measured quantitative parameters, or defining threshold judgments for qualitative status changes (e.g., platelet activation, etc.). For example, Pivkin et al.59 treated platelets as having three possible biological states: passive and non-adhesive, triggered, and activated and adhesive. However, as mentioned above, non-activated platelets also interact with injured vessel walls and translocation cannot be recovered in this model.
Mori et al.53 applied Voigt’s model to calculate the binding force between platelets, where the aggregation of platelets is mediated by both vWF and fibrinogen. In their model, the association of GPIIb/IIIa–fibrinogen–GPIIb/IIIa and GPIbα–vWF–GPIbα between two platelets is determined by judging the difference in the platelets’ velocities. The binding force is unbiased for the possible receptor configurations on the cell membrane between two platelets.
On the other hand, Monte Carlo simulation of receptor–ligand binding between surfaces (known as adhesive dynamics, or AD)25 treats individual receptors on the cell membrane as separate units and the rate of formation/breakage of each receptor–ligand bond is determined by the Bell Model.5 The Bell Model calculates the probability of the dissociation event occurring over a specific timespan based on the stretched distance between the roots of the target receptor and the ligand, which is deviated from the relaxed form. As a result, AD can recover complex cell–cell interaction behavior, including translocation, temporary tethering and adhesion. In 1992, AD succeeded in simulating the rolling and adhesion of neutrophils mediated by CD62/LECAM-1.25 AD was later applied to reveal the hydrodynamic interaction between stably rolling cells, and between rolling and flowing cells as well.34,35 AD was first applied to platelets in 2007, where Mody and King49 assessed the influence of Brownian motion on platelet transport and single-bond adhesion behavior near wall. AD also successfully recovered the shear-induced platelet aggregation mediated by GPIbα–vWF bridging.50
It is interesting to note that, though computational models are constantly being improved, experimental results that assign measured values to model parameters are sometimes prone to revision as well. For instance, the dissociation kinetics of the platelet GPIbα–vWF bond was first determined to rely on the Bell Model, which posits a “slip-bond.”14 The unstressed dissociation constant was also determined by a cell-tethering assay.14 Later, optical tweezer techniques were applied to single-molecule experiments to obtain a more precise measurement of the dissociation constant.2 In 2008, researchers discovered that the dissociation of GPIbα–vWF demonstrates a “catch-bond” regime, similar to that of selectins.10,81 To reflect the “catch-bond” properties, the governing kinetic equations were modified from the original Bell Model.4 In 2010, a novel experimental technique called ReaLiSM was described, which allows repeated measurements of the binding and unbinding of a receptor and ligand in a single molecule. ReaLiSM revealed that, instead of “slip-bond” and “catch-bond” transition, GPIbα–vWF has two “slip-bond” regimes, termed a “flex-bond.”33 Though it is still a controversial topic, it is important to be aware of possible changes in the currently accepted experimental data, which serves as the most convincing method of validation for simulation studies.
Platelet Activation
Platelet activation is also an essential process during hemostasis. Numerous studies have demonstrated that when a resting platelet encounters agonists such as collagen, thrombin, thromboxane, or ADP, it undergoes a change in shape (from ellipsoid to spheroid), degranulation and becomes fully adherent.86 Munnix et al.56 summarized current evidence of the heterogeneity of platelet response to various environmental factors and intrinsic differences (e.g., protein expression level, platelet aging) during thrombus formation. They classified deposited platelets inside a thrombus into three subpopulations, namely “aggregating platelets with reversible integrin activation,” “procoagulant (coated) platelets exposing phosphatidylserine and binding coagulation factors,” and “contracting platelets with cell–cell contacts,” with each subpopulation possessing unique morphology, surface characteristics and functionality.56 Heterogeneity is also exhibited by suspended platelets in the plasma as well.56 Purvis et al.63,64 developed a molecularly detailed, bottom-up ordinary differential equation (ODE) model that simulates ADP-mediated activation. Sorensen et al.69,70 utilized a coupled set of convection–diffusion-reaction equations to describe thrombin-induced platelet activation and deposition. These platelet activation models have not yet been combined with models, but there is growing expectation that they should be included in thrombus growth simulations because of the significant behavioral differences between non-activated and activated platelets.
Cell Mechanics
In different computational models, the mechanical properties of platelets are usually approximated in one way or another: platelets are treated either as possessing a spherical shape, solid texture, uniform size distribution, or any combination of these. The size of a platelet, commonly represented by the variable mean platelet volume, was found to be associated with a higher risk of thrombosis, stenosis, and cardiovascular disease.9 The shape factor of platelets has also been shown to be associated with normal platelet function, and the resulting transient tethering due to simple-shear flow has been explored.51,52 While it is relatively easy to adjust the size and shape of a platelet in simulations, it is somewhat more challenging to incorporate realistic deformability of these cells into the model. Since the stiffness of blood cells greatly influences a cell’s ability to marginate76 and the mechanical properties of the encompassing thrombus, it is important to not only consider the other blood constituents with relatively large volume fractions (e.g., RBCs), but also to treat them as deformable particles. Pozrikidis developed the membrane constitutive equations for deformable RBCs.62 Membrane in-plane tension, transverse shear tension, and membrane bending moments are added to the cell membrane force and torque balance to conserve the deformable biconcave shape of RBCs61 and the cell volume. Based on Pozrikidis’s membrane mechanics, Zhao et al.83 established a spectral boundary integral method that is able to simulate deformable RBCs flowing inside vessels. An similar approach can also be applied to simulate a deformable thrombus or an individual platelet.
The structural properties and contractile force of a clot have been known for decades.29 Recently, single platelet mechanics and contraction dynamics were experimentally determined by atomic force microscopy, which can provide insights into the clot stiffening process.38 Experimentally determined mechanical parameters of platelets and thrombus provide solid background for simulation studies.
Platelet Adhesive Dynamics
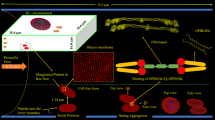
Platelet Adhesive Dynamics, defined by Mody and King,50,51 combines a Monte Carlo simulation of receptor–ligand binding (i.e., Adhesive Dynamics, or AD) and hydrodynamic calculations with CDL-BIEM. This model has been applied to the scenario where ellipsoid platelets are flowing/tethering close to the vessel wall. The study of platelet aggregation under high shear via GPIbα–vWF–GPIbα bridging contained details such as the physical structure of large multimeric protein vWF (Fig. 2a). The PAD model successfully explained the mechanisms of Type 2 von Willibrand Disease (vWD), where the subgroups of either large or small vWF multimers are absent. Mody and King discussed that an increase in vWF ligand size in plasma strongly influences the equilibrium binding kinetics and correspondingly increases platelet–platelet binding efficiency. PAD was also able to explore the complex and nonlinear force loading curve for each individual GPIbα–vWF–GPIbα bond during two-platelet aggregation. Recently, PAD demonstrated the periodic translocating motion of a platelet on an injured vessel surface, where a fast flipping period and a slow tilting period were shown for an ellipsoid-shaped platelet (Figs. 2b, 2c). By combining with other type of receptor–ligand binding pairs such as integrin α2β1–collagen, and αIIbβ3–fibrinogen, PAD will be able to recover the transition from translocation on the sub-endothelial surface to firm adhesion, as well as platelet aggregation after activation. Thus, PAD has the potential to reveal the complete biophysical role of platelets during hemostasis/thrombosis.

(a) Schematic diagram of the platelet–platelet bridging that depicts two platelets transiently aggregating in linear shear flow via GPIbα–vWF–GPIbα bonds. (b) Diagram showing translocation of a platelet under linear shear flow via repeated bond dissociation from the upstream side and bond formation at the downstream side. (c) The linear velocity of the center of the platelet at a shear rate of 1250 s−1. The components of the depicted diagram are not drawn to scale
Future Directions
While increasing knowledge of biochemical/biophysical mechanisms of hemostasis and thrombosis has been revealed, computational models are being built towards more comprehensive settings to simulate more realistic physiological processes. However, such model improvement often prohibitively increases the consumption of computer processing power. For instance, the simulation of two platelets aggregating in 3D, which is provided by PAD, can run for days in order to obtain a few seconds of real-time behavior. Even with the rapid evolution of new generations of CPUs, as well as the development of parallel computing algorithms applied on computer arrays, simulations are usually time consuming and computationally expensive. The coarse-grained approach simplifies the computing methods while preserving the fidelity of the model. Coarse-grained methods are also very understandable and easily transferrable, and thus enable more broad collaboration. Though they may be subject to accuracy concerns, it is expected that they will be an important approach and will be incorporated into a variety of models.
While most multiscale simulation models are still prone to verify an identified physiological mechanism (by which they validate themselves at the same time), researchers’ original motivation for developing simulations is their potential predictive power. Mody and King50 applied simulations and predicted that the 2-D cross-linking rate of the GPIbα–vWF bond appears to be consistent with a piecewise linear model dependent on the imposed fluid shear rate, with a weak dependence on shear rate followed by a stronger dependence on shear rate. Later, Singh et al.68 experimentally presented a similar piecewise kinetics dependence for vWF-bisANS binding, proposing the unfolding mechanism of vWF under shear flow. Another example of model prediction is the systems biology approach of S. Diamond and colleagues comprised of a complete bottom-up simulation of platelet metabolism based on a Monte Carlo algorithm which is reaching a point where patient-specific prediction of coagulation kinetics may be possible.13
Multiscale simulations reinforce the significance of in vitro studies by filling the gaps between in vitro experiments to in vivo human thrombus formation. While it is difficult to recover the full spectrum of in vivo conditions in vitro, ideally conditioned in vitro experiments provide accurate measurement of chemical/physical parameters. Those parameters can be incorporated into models to better recreate realistic and complex in vivo conditions. On the other hand, the quality of simulations is heavily dependent on the quality of in vitro data, as the accuracy of the model parameter measurements is critical to support the fidelity of simulation models.
It is hoped that the employment of multiscale simulations of hemostasis and thrombosis will be focused towards clinical predictions, such as diagnosis of a stenosis configuration, estimation of its progression rate and prognosis after surgery. Simulations may also facilitate personalized medicine. An example of this might be a patient with a femoral artery stenosis being implanted with a stent whose geometry is specifically designed to reduce the possibility of turbulent and high-shear flow, which will help prevent further thrombosis in the narrowing vessel region. Such stent design can be supported by recovery of the 3D structure of the patient’s femoral artery through non-invasive imaging, followed by CFD calculation of the associated fluid mechanics. Another example which focuses on the microscopic level is specifically designed small soluble protein molecules introduced into the blood circulation system, where the molecule is capable of restoring normal vWF function in type 1 von Willebrand Disease patients. Such protein design may be based on patient-specific genome sequencing, vWF structure recovery and MD simulation with thousands of small molecule candidate compounds. Simulation might also predict the physiological effects where the pharmacodynamics of a drug can be individually determined for each patient beforehand.
Future simulations of the hemostasis process may also involve other physiological processes. Platelets, for example, were long thought to associate with cancer metastasis since the early 1990s.26 More recently, it has been well studied that platelet activation and blood coagulation have a crucial role in the progression of cancer.24 Platelets also physically promote tumor metastasis by protecting tumor cells from immune elimination and enhancing their arrest on the endothelium.24 By introducing cancer cells into hemostasis models, researchers will be able to simulate platelet-mediated tumor metastasis. Platelets also play other pathological roles in inflammation and atherogenesis.23 In summary, multiscale simulations of hemostasis will be more versatile in the future, being applied in different biological fields.
Code Sharing
Computer simulation of biological processes has expanded tremendously over the past two decades. Storage databases, such as those used for genomic data in computational genetics, have been widely shared. However, databases of methods (i.e., algorithms or modules) have not seen the same pooling of resources. While commercially available packages are costly and inflexible toward adjustment, highly compatible platforms have been established for better communication, wider application and more powerful combined modules. Those platforms have a list of compatible components from markup languages and simulation tools to model databases and discussion forums. System Biology Markup Language (SBML) and one of its derivatives, CellML, are examples of common tools for multiple-scale analysis and simulation of biological systems, with the latter placing a special emphasis on cellular phenomena. SBML and CellML are powerful for simulating the coagulation cascade and platelet activation, but are not optimal for physical models, such as cell motion or deformation under flow. Closer collaboration will be required in order to develop comprehensive and more realistic models. Working groups have been formed in order to better serve the networking purposes of computational biologists; these also facilitate the sharing of codes that are not readily transferrable to common platforms. The Multiscale System Biology Working Group is such an example.27 The group establishes a website platform with up-to-date information on available tools, novel concepts, and major, relevant review papers in the field. The goal is to identify and articulate current challenges and opportunities in the field, as well as to foster scientific collaboration.
References
Anderson, J. D. Computational Fluid Dynamics: The Basics with Applications. McGraw-Hill Series in Mechanical Engineering. New York: McGraw-Hill, p. xxiv, 547 pp., 1995.
Arya, M., B. Anvari, G. M. Romo, M. A. Cruz, J. F. Dong, L. V. McIntire, J. L. Moake, and J. A. Lopez. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib–IX complex: studies using optical tweezers. Blood 99(11):3971–3977, 2002.
Ataullakhanov, F. I., and M. A. Panteleev. Mathematical modeling and computer simulation in blood coagulation. Pathophysiol. Haemost. Thromb. 34(2–3):60–70, 2005.
Auton, M., C. Zhu, and M. A. Cruz. The mechanism of VWF-mediated platelet GPIb alpha binding. Biophys. J. 99(4):1192–1201, 2010.
Bell, G. I. Models for the specific adhesion of cells to cells. Science 200(4342):618–627, 1978.
Bennett, J. S. Structure and function of the platelet integrin alphaIIbbeta3. J. Clin. Investig. 115(12):3363–3369, 2005.
Brady, J. F., and G. Bossis. Stokesian dynamics. Annu. Rev. Fluid Mech. 20:111–157, 1988.
Chatterjee, M. S., W. S. Denney, H. Y. Jing, and S. L. Diamond. Systems biology of coagulation initiation: kinetics of thrombin generation in resting and activated human blood. PLoS Comput. Biol. 6(9):e1000950, 2010.
Chu, S. G., R. C. Becker, P. B. Berger, D. L. Bhatt, J. W. Eikelboom, B. Konkle, E. R. Mohler, M. P. Reilly, and J. S. Berger. Mean platelet volume as a predictor of cardiovascular risk: a systematic review and meta-analysis. J. Thromb. Haemost. 8(1):148–156, 2010.
Coburn, L. A., V. S. Damaraju, S. Dozic, S. G. Eskin, M. A. Cruz, and L. V. McIntire. GPIb alpha-vWF rolling under shear stress shows differences between type 2B and 2M von Willebrand disease. Biophys. J. 100(2):304–312, 2011.
Colman, R. W. Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins, p. xxiv, 1827 pp., 2006.
Davi, G., and C. Patrono. Platelet activation and atherothrombosis. N. Engl. J. Med. 357(24):2482–2494, 2007.
Diamond, S. L. Systems biology to predict blood function. J. Thromb. Haemost. 7:177–180, 2009.
Doggett, T. A., G. Girdhar, A. Lawshe, D. W. Schmidtke, I. J. Laurenzi, S. L. Diamond, and T. G. Diacovo. Selectin-like kinetics and biomechanics promote rapid platelet adhesion in flow: the GPIb(alpha)-vWF tether bond. Biophys. J. 83(1):194–205, 2002.
Einav, S., and D. Bluestein. Dynamics of blood flow and platelet transport in pathological vessels. Cardiac engineering: from genes and cells to structure and function. Ann. N. Y. Acad. Sci. 1015:351–366, 2004.
Filipovic, N., M. Kojic, and A. Tsuda. Modelling thrombosis using dissipative particle dynamics method. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 366(1879):3265–3279, 2008.
Fogelson, A. L. Continuum models of platelet-aggregation—formulation and mechanical-properties. Siam J. Appl. Math. 52(4):1089–1110, 1992.
Fogelson, A. L., and R. D. Guy. Platelet-wall interactions in continuum models of platelet thrombosis: formulation and numerical solution. IMA Math. Med. Biol. J. 21(4):293–334, 2004.
Fogelson, A. L., Y. H. Hussain, and K. Leiderman. Blood clot formation under flow: the importance of factor XI depends strongly on platelet count. Biophys. J. 102(1):10–18, 2012.
Fogelson, A. L., and J. P. Keener. Toward an understanding of fibrin branching structure. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 81(5 Pt 1):051922, 2010.
Fogelson, A. L., and N. Tania. Coagulation under flow: the influence of flow-mediated transport on the initiation and inhibition of coagulation. Pathophysiol. Haemost. Thromb. 34(2–3):91–108, 2005.
Gailani, D., and T. Renne. Intrinsic pathway of coagulation and arterial thrombosis. Arterioscler. Thromb. Vasc. Biol. 27(12):2507–2513, 2007.
Gawaz, M., H. Langer, and A. E. May. Platelets in inflammation and atherogenesis. J. Clin. Investig. 115(12):3378–3384, 2005.
Gay, L. J., and B. Felding-Habermann. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 11(2):123–134, 2011.
Hammer, D. A., and S. M. Apte. Simulation of cell rolling and adhesion on surfaces in shear flow: general results and analysis of selectin-mediated neutrophil adhesion. Biophys. J. 63(1):35–57, 1992.
Honn, K. V., D. G. Tang, and J. D. Crissman. Platelets and cancer metastasis—a causal relationship. Cancer Metastasis Rev. 11(3–4):325–351, 1992.
http://www.imagwiki.nibib.nih.gov/mediawiki/index.php?title=Multiscale_Systems_Biology_Working_Group
Hyakutake, T., T. Matsumoto, and S. Yanase. Lattice Boltzmann simulation of blood cell behavior at microvascular bifurcations. Math. Comput. Simul. 72(2–6):134–140, 2006.
Jen, C. J., and L. V. Mcintire. The structural-properties and contractile—force of a clot. Cell Motil. Cytoskelet. 2(5):445–455, 1982.
Jones, K. C., and K. G. Mann. A model for the tissue factor pathway to thrombin. 2. A mathematical simulation. J. Biol. Chem. 269(37):23367–23373, 1994.
Jung, S. M., and M. Moroi. Activation of the platelet collagen receptor integrin alpha(2)beta(1): its mechanism and participation in the physiological functions of platelets. Trends Cardiovasc. Med. 10(7):285–292, 2000.
Kim, S., and S. J. Karrila. Microhydrodynamics: Principles and Selected Applications. Butterworth-Heinemann Series in Chemical Engineering. Boston: Butterworth-Heinemann, p. xxiii, 507 pp., 1991.
Kim, J., C. Z. Zhang, X. H. Zhang, and T. A. Springer. A mechanically stabilized receptor–ligand flex-bond important in the vasculature. Nature 466(7309):992–995, 2010.
King, M. R., and D. A. Hammer. Multiparticle adhesive dynamics: hydrodynamic recruitment of rolling leukocytes. Proc. Natl Acad. Sci. USA 98(26):14919–14924, 2001.
King, M. R., and D. A. Hammer. Multiparticle adhesive dynamics. Interactions between stably rolling cells. Biophys. J. 81(2):799–813, 2001.
Kroll, M. H., T. S. Harris, J. L. Moake, R. I. Handin, and A. I. Schafer. von Willebrand factor binding to platelet GpIb initiates signals for platelet activation. J. Clin. Investig. 88(5):1568–1573, 1991.
Kuharsky, A. L., and A. L. Fogelson. Surface-mediated control of blood coagulation: the role of binding site densities and platelet deposition. Biophys. J. 80(3):1050–1074, 2001.
Lam, W. A., O. Chaudhuri, A. Crow, K. D. Webster, T. D. Li, A. Kita, J. Huang, and D. A. Fletcher. Mechanics and contraction dynamics of single platelets and implications for clot stiffening. Nat. Mater. 10(1):61–66, 2011.
Laurens, N., P. Koolwijk, and M. P. de Maat. Fibrin structure and wound healing. J. Thromb. Haemost. 4(5):932–939, 2006.
Leiderman, K., and A. L. Fogelson. Grow with the flow: a spatial-temporal model of platelet deposition and blood coagulation under flow. IMA Math. Med. Biol. J. 28(1):47–84, 2011.
Lippi, G., E. J. Favaloro, M. Franchini, and G. C. Guidi. Milestones and perspectives in coagulation and hemostasis. Semin. Thromb. Hemost. 35(1):9–22, 2009.
Lo, K., W. S. Denney, and S. L. Diamond. Stochastic modeling of blood coagulation initiation. Pathophysiol. Haemost. Thromb. 34(2–3):80–90, 2005.
Luan, D., F. Szlam, K. A. Tanaka, P. S. Barie, and J. D. Varner. Ensembles of uncertain mathematical models can identify network response to therapeutic interventions. Mol. BioSyst. 6(11):2272–2286, 2010.
Luan, D., M. Zai, and J. D. Varner. Computationally derived points of fragility of a human cascade are consistent with current therapeutic strategies. PLoS Comput. Biol. 3(7):e142, 2007.
Mackman, N., R. E. Tilley, and N. S. Key. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 27(8):1687–1693, 2007.
MacMeccan, R. M., J. R. Clausen, G. P. Neitzel, and C. K. Aidun. Simulating deformable particle suspensions using a coupled lattice-Boltzmann and finite-element method. J. Fluid Mech. 618:13–39, 2009.
Merrill, E. W. Rheology of blood. Physiol. Rev. 49(4):863–868, 1969.
Mody, N. A., and M. R. King. Three-dimensional simulations of a platelet-shaped spheroid near a wall in shear flow. Phys. Fluids 17(11):113302, 2005.
Mody, N. A., and M. R. King. Influence of Brownian motion on blood platelet flow behavior and adhesive dynamics near a planar wall. Langmuir 23(11):6321–6328, 2007.
Mody, N. A., and M. R. King. Platelet adhesive dynamics. Part II: high shear-induced transient aggregation via GPIbalpha–Vwf–GPIbalpha bridging. Biophys. J. 95(5):2556–2574, 2008.
Mody, N. A., and M. R. King. Platelet adhesive dynamics. Part I: characterization of platelet hydrodynamic collisions and wall effects. Biophys. J. 95(5):2539–2555, 2008.
Mody, N. A., O. Lomakin, T. A. Doggett, T. G. Diacovo, and M. R. King. Mechanics of transient platelet adhesion to von Willebrand factor under flow. Biophys. J. 88(2):1432–1443, 2005.
Mori, D., K. Yano, K. Tsubota, T. Ishikawa, S. Wada, and T. Yamaguchi. Simulation of platelet adhesion and aggregation regulated by fibrinogen and von Willebrand factor. Thromb. Haemost. 99(1):108–115, 2008.
Mori, D., K. Yano, K. Tsubota, T. Ishikawa, S. Wada, and T. Yamaguchi. Computational study on effect of red blood cells on primary thrombus formation. Thromb. Res. 123(1):114–121, 2008.
Munn, L. L., and M. M. Dupin. Blood cell interactions and segregation in flow. Ann. Biomed. Eng. 36(4):534–544, 2008.
Munnix, I. C. A., J. M. E. M. Cosemans, J. M. Auger, and J. W. M. Heemskerk. Platelet response heterogeneity in thrombus formation. Thromb. Haemost. 102(6):1149–1156, 2009.
Murata, T. Theory of non-newtonian viscosity of red-blood-cell suspension—effect of red-cell deformation. Biorheology 20(5):471–483, 1983.
Phan-Thien, N., D. Tullock, and S. Kim. Completed double-layer in half-space: a boundary element method. Comput. Mech. 9:121–135, 1992.
Pivkin, I. V., P. D. Richardson, and G. Karniadakis. Blood flow velocity effects and role of activation delay time on growth and form of platelet thrombi. Proc. Natl Acad. Sci. USA 103(46):17164–17169, 2006.
Pivkin, I. V., P. D. Richardson, and G. E. Karniadakis. Effect of red blood cells on platelet aggregation. IEEE Eng. Med. Biol. Mag. 28(2):32–37, 2009.
Pozrikidis, C. Numerical simulation of the flow-induced deformation of red blood cells. Ann. Biomed. Eng. 31(10):1194–1205, 2003.
Pozrikidis, C. Axisymmetric motion of a file of red blood cells through capillaries. Phys. Fluids 17(3):031503, 2005.
Purvis, J. E., M. S. Chatterjee, L. F. Brass, and S. L. Diamond. A molecular signaling model of platelet phosphoinositide and calcium regulation during homeostasis and P2Y1 activation. Blood 112(10):4069–4079, 2008.
Purvis, J. E., R. Radhakrishnan, and S. L. Diamond. Steady-state kinetic modeling constrains cellular resting states and dynamic behavior. PLoS Comput. Biol. 5(3):e1000298, 2009.
Rayz, V. L., L. Boussel, L. Ge, J. R. Leach, A. J. Martin, M. T. Lawton, C. McCulloch, and D. Saloner. Flow residence time and regions of intraluminal thrombus deposition in intracranial aneurysms. Ann. Biomed. Eng. 38(10):3058–3069, 2010.
Sakariassen, K. S., P. A. Bolhuis, and J. J. Sixma. Human blood platelet adhesion to artery subendothelium is mediated by factor VIII-Von Willebrand factor bound to the subendothelium. Nature 279(5714):636–638, 1979.
Savage, B., E. Saldivar, and Z. M. Ruggeri. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 84(2):289–297, 1996.
Singh, I., E. Themistou, L. Porcar, and S. Neelamegham. Fluid shear induces conformation change in human blood protein von Willebrand factor in solution. Biophys. J. 96(6):2313–2320, 2009.
Sorensen, E. N., G. W. Burgreen, W. R. Wagner, and J. F. Antaki. Computational simulation of platelet deposition and activation: I. Model development and properties. Ann. Biomed. Eng. 27(4):436–448, 1999.
Sorensen, E. N., G. W. Burgreen, W. R. Wagner, and J. F. Antaki. Computational simulation of platelet deposition and activation: II. Results for Poiseuille flow over collagen. Ann. Biomed. Eng. 27(4):449–458, 1999.
Steinman, D. A. Image-based computational fluid dynamics modeling in realistic arterial geometries. Ann. Biomed. Eng. 30(4):483–497, 2002.
Tokarev, A. A., A. A. Butylin, E. A. Ermakova, E. E. Shnol, G. P. Panasenko, and F. I. Ataullakhanov. Finite platelet size could be responsible for platelet margination effect. Biophys. J. 101(8):1835–1843, 2011.
Undas, A., and R. A. S. Ariens. Fibrin clot structure and function a role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler. Thromb. Vasc. Biol. 31(12):E88–E99, 2011.
Wang, N. T., and A. L. Fogelson. Computational methods for continuum models of platelet aggregation. J. Comput. Phys. 151(2):649–675, 1999.
Weisel, J. W. Structure of fibrin: impact on clot stability. J. Thromb. Haemost. 5(Suppl 1):116–124, 2007.
Wiggs, B. R., D. English, W. M. Quinlan, N. A. Doyle, J. C. Hogg, and C. M. Doerschuk. Contributions of capillary pathway size and neutrophil deformability to neutrophil transit through rabbit lungs. J. Appl. Physiol. 77(1):463–470, 1994.
Wu, J. S., and C. K. Aidun. Simulating 3D deformable particle suspensions using lattice Boltzmann method with discrete external boundary force. Int. J. Numer. Methods Fluids 62(7):765–783, 2010.
Xu, Z., N. Chen, M. M. Kamocka, E. D. Rosen, and M. Alber. A multiscale model of thrombus development. J. R. Soc. Interface 5(24):705–722, 2008.
Xu, Z., M. Kamocka, M. Alber, and E. D. Rosen. Computational approaches to studying thrombus development. Arterioscler. Thromb. Vasc. Biol. 31(3):500–505, 2011.
Xu, Z. L., J. H. Lioi, J. A. Mu, M. M. Kamocka, X. M. Liu, D. Z. Chen, E. D. Rosen, and M. Alber. A multiscale model of venous thrombus formation with surface-mediated control of blood coagulation cascade (vol 98, pg 1723, 2010). Biophys. J. 99(7):2384–2385, 2010.
Yago, T., J. Lou, T. Wu, J. Yang, J. J. Miner, L. Coburn, J. A. Lopez, M. A. Cruz, J. F. Dong, L. V. McIntire, R. P. McEver, and C. Zhu. Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J. Clin. Investig. 118(9):3195–3207, 2008.
Yamaguchi, T., T. Ishikawa, Y. Imai, N. Matsuki, M. Xenos, Y. F. Deng, and D. Bluestein. Particle-based methods for multiscale modeling of blood flow in the circulation and in devices: challenges and future directions. Ann. Biomed. Eng. 38(3):1225–1235, 2010.
Zhao, H., A. H. G. Isfahani, L. N. Olson, and J. B. Freund. A spectral boundary integral method for flowing blood cells. J. Comput. Phys. 229(10):3726–3744, 2010.
Zhao, R., M. V. Kameneva, and J. F. Antaki. Investigation of platelet margination phenomena at elevated shear stress. Biorheology 44(3):161–177, 2007.
Zhao, H., E. S. G. Shaqfehhear, and V. Narsimhan. Shear-induced particle migration and margination in a cellular suspension. Phys. Fluids 24:011902, 2012.
Zucker, M. B., and V. T. Nachmias. Platelet activation. Arteriosclerosis 5(1):2–18, 1985.
Acknowledgments
The authors thank John P. Lindsey for discussions and review of the manuscript. The work is funded by NIH Grant No. HL097971.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Scott L. Diamond oversaw the review of this article.
Rights and permissions
About this article
Cite this article
Wang, W., King, M.R. Multiscale Modeling of Platelet Adhesion and Thrombus Growth. Ann Biomed Eng 40, 2345–2354 (2012). https://doi.org/10.1007/s10439-012-0558-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-012-0558-8