
Abstract
In the present study, we investigated the genetic structure and diversity of P. nigra populations in Bulgaria, using simple sequence nuclear repeats. Among-population structure was studied with distance and Bayesian frequency methods, assuming geometric distance and a “non-admixture” model. The “NJ” and “non-admixture” clusters confirm the “mountain effect” hypothesis of the black pine genetic structure in the study region. The analyses showed moderate among-population divergence (13.31 %; AMOVA) and evidence of genetic bottlenecks. The coalescent analyses suggest that P. nigra has survived for a long period (thousands of generations) under strong selection pressure and that its populations continued to be exposed to stochastic factors like climate fluctuation, forest fire and disease. The combination of recent and historic changes is responsible for the present population size and genetic diversity. Our results suggest that conservation and management practices should strive to maintain this genetic differentiation, specifically by emphasising reforestation efforts with stocks from local provenances to avoid non-local introductions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The assessment of population genetic diversity is important for building the strategy for the conservation of threatened and endangered species. The significant reduction in the genetic diversity for species with a widespread distribution and high commercial pressure is often underestimated. The importance of maintaining a high level of genetic diversity for populations in constant anthropogenic and ecological pressures has long been debated (Lehman 1998). While it is well known that genetic diversity is important for adaptation and speciation, it is less evident that how low diversity, as detected by molecular loci, impacts upon the capacity of populations to survive in the long term (Willi et al. 2006; Pertoldi et al. 2007). The limits of population diversity depend on many factors, such as fluctuation in population size and structure at the meta- and regional geographic levels (Pimm et al. 1989; Hedrick 1996). The recent and severe reduction in population sizes in general may have caused the loss a significant level of genetic diversity due to drift and inbreeding depression. The management and conservation practices are therefore important to understand how the populations can adapt to a reduction in size and the potential genetic consequences that it may produce. These answers will largely depend on their history (Keller and Waller 2002; Leberg and Firmin 2008).
Population management is important for forest tree populations and species and is a pressing challenge. Forest trees are the foundation of an ecosystem in which various plants, insects and animals are closely interrelated. The situation is particularly acute for populations from the glacial refugium in the southern part of Europe. These populations carry a major part of genetic diversity and are usually over-exploited during periods of economic crisis, carrying with it the concomitant risk of pauperisation and dramatic extinction. The European black pine (genus Pinus; P. nigra Arn.) in the Balkan Peninsula falls into this category. The privatisation of forest land in the last 20 years has caused a severe decline in some populations where European black pine grows naturally. This pine has been one of the major species used for reforestation in sub-Mediterranean region. It exhibits high phenotypic variability in terms of its morphological, anatomical and physiological characteristics (Vidakovic 1974, 1991; Scaltsoyiannes et al. 1994a, b).
In a previous study, we investigated chloroplast diversity and terpene content in P. nigra provenance tests in Bulgaria (Naydenov et al. 2006): it was the first study of chloroplast DNA variations for European black pine. Variation in cpSSR (chloroplast simple sequence repeats) separated P. nigra into four groups, and the pattern of inter-population variability suggested that provenances from the Eastern Rhodope, Sr. Gora (Sredna Gora) and St. Planina (Stara Planina) Mountains have a common postglacial migration history different from that of other provenances. We were also able to distinguish four groups in Bulgaria, using terpene analysis. The two principal differences between the results of cpSSR and terpene analyses, in relation to among-population variability, are (1) divergence between marginal populations and (2) group fragmentation in the Rhodope/Pirin region. The discrepancy in among-population structure revealed between terpene and cpSSR analysis in the same area is perhaps a result of more complicated and different migration history of pollen vs seed and different patterns of heredity in used markers. Further studies should be completed to ensure that the genetic structure of P. nigra is understood throughout Bulgaria, so that continental planting can be undertaken in a way that ensures continuity of genetic structure and diversity of this species.
We investigated genetic variations at 12 nuclear microsatellite loci among nine populations from provenance tests that had been gathered 30 years ago in Bulgaria. The first hypothesis suggests that it is possible to demonstrate the mountain origin—“mountain effect” from its population structure, i.e. “mountain effect”. This is a term describing the distinct genetic structure specific for each mountain. It was only possible to observe the recent reduction in effective population size for the second hypothesis. We were seeking to find out whether the level of diversity is due to recent or to more historic changes in population size. The results from this study are the first, according to our knowledge, and they have important implications for the utility of provenance tests of forest trees and for conservation and management practices in Bulgaria.
Materials and methods
Plant material
The plant material consisted of different populations: three populations are from the Rhodope Mountains/S–W Bulgaria (Pops-A, C and D); and one each from the Rila/S–W Bulgaria (Pop-B), St. Planina/Central Bulgaria (Pop-E), Sr. Gora/Central Bulgaria (Pop-F), Slavianka/S–W Bulgaria (Pop-G), Pirin/S–W Bulgaria (Pop-H) and Osogovo/W Bulgaria (Pop-I) ranges. A total of 324 trees from the nine populations (36 × 9) represented a very intensive sampling effort for the natural distribution of P. nigra over a region covering more than 44,000 km2 and a typical altitudinal range of 600–1100 m (Table 1; Fig. 1). The collected seeds were individually separated and placed in Petri dishes at 26 °C and under a 14-h light regime for 2 days.

Locations of the original populations of the investigated provenances. Pop-A (Raven), Pop-C (Sarnica) and Pop-D (Momchilovci) are from the Rhodope Mountain; Pop-B (Belovo) is from the north-eastern portion of the Rila Mountain; Pop-E (Chipca) is from the central portion of St. Planina Mountain; Pop-F (Ch. Danovo) is from the central portion of Sr. Gora Mountain; Pop-G (Katunci) is from the north-western portion of the Slavianka Mountain; Pop-H (Dobrinishte) is from the north-eastern portion of the Pirin Mountain; and Pop-I (Gabra) is from the Osogovo Mountain (left). For natural distribution of European black pine in Bulgaria (right top) and Europe (right bottom), please see www.euforgen.org/distribution_maps.html
The plant material was collected for a large provenance test following the techniques similar to those described by Naydenov et al. (2006). Briefly, the provenance test “Sirakovo” was created 30 years ago with the seedlings from first selection stepmother trees from different provenances. The mother trees were chosen following the instructions of the Forest service of Bulgaria, IUFRO, FAO and USDA as described in Naydenov et al. (1993/1996, 2006) with a minimum distance between each tree of 50 m. A strict protocol concerning seed management was implemented for a period of 5 years. This ensured that there was no mixing of seed provenances and that they were properly identified. The protocol was in place for the entire 5-year period of seed collection, seedling production and provenance test establishment. Each mother tree from a distinct provenance is represented in the provenance tests by one individual in each block of repetition. Only healthy seedlings were used there, i.e. without visible damage from insect attacks or disease. No special pre-selection screenings (by colour or dimension) were applied to choose seedlings for the provenance test. In this provenance test, we have 12 blocks of repetitions per provenance; each block has 64 trees representing distinct mother trees from one provenance. We sampled one block per provenance to avoid sampling the same mother tree twice and to avoid artificially increasing inbreeding scores by mishandling. Healthy seeds were gathered from nine populations of P. nigra which were situated in the continental European climatic zone, Low Mountain sub-region of Bulgaria (Balkan Peninsula), according to the climatic map of Bondev (1973). From each individual, 10–20 g of seeds was collected and stored separately in the dark at 4 °C. Thirty-six healthy individuals that were free of disease and insect attack were sampled from each provenance.
Nuclear DNA: laboratory methods
Total DNA was extracted from 36 trees/population using the genomic DNA mini-preparation kit (Sigma# G2N350). From each individual, between 10 and 12 megagametophytes were used to determine its genetic profile. Genotyping of individuals was performed by screening for 12 nuclear primer pairs originally developed for other Pinus species: Pde-3, Pde-5, Pde-7, PtTX-2090, PtTX-2123, PtTX-2146, PtTX-2158, PtTX-3003, PtTX-3020, PtTX-3025, PtTX-3030 and PtTX-3118 (Lian et al. 2000; Auckland et al. 2002). The 25–35 ng of template DNA was used in PCR master mix containing 50 mM Tris buffer (pH 9.0), 20 mM ammonium sulphate, 100 μg/μL gelatine, 1 % DMSO, 50 μM of each dNTP, 100 nM of forward and reverse primer, 0.325 U HotMasterHot Master Taq DNA Polymerase (Eppendorf AG) and 1.5–4.0 mM MgCl2 in a 10-µl reaction volume. Loci used in this analysis were amplified in a thermo-cycler (Perkin-Elmer 9700) under the following protocol: initial 2.30-min Taq activation at 95 °C; 30 cycles consisting of 25-s denaturation at 94 °C, 20-s annealing temperature optimised for each primer set and 1-min extension temperature at 72 °C; final extension for 3.30 min at 72 °C. The PCR analysis was similar to that in Williams et al. (2000).
After initial amplification, the PCR product was diluted by 5–8 times. Fluorescent dye-labelled PCR products (0.65 µl), 12 µl Hi-Di formamide (Applied Biosystems) and internal size standard 0.36 µl TAMRA-500 (Applied Biosystems) were denatured for 5 min at 95 °C, placed on ice and separated by capillary electrophoresis on an Applied Biosystems Prism 310 Genetic Analyser. The data were analysed using Genscan™ software (Applied Biosystems), and the genotypes of the samples were inferred from the individual size profiles of the nuclear DNA markers using GeneMAPPER ver. 3.7 software (Applied Biosystems). The MicroCHECKER ver. 2.2.3 program of Van Oosterhout et al. (2004) was used to test for “null” alleles and scoring errors caused by large allele dropouts and stutter peaks.
Nuclear DNA: statistical analysis
The respective gene diversity F- and G-statistics of Wrigth (1965) and Nei (1975) were calculated using GENETIX V.4.02 (Belkhir 2002), ARLEQUIN V.3.001 (Excoffier et al. 2002) and GENALEX V.6 (Peakall and Smouse 2006). The effective number of alleles (A e) and private alleles (A pa) was estimated according to Shannon (1948a, b) and Kimura and Crow (1964). Symmetric gene flow (N m) was estimated as the product of the proportion of migrants and effective population size. Estimation of N m was based on the fixation index F st (N m = (1 − F st)/4F st; Slatkin 1987; Slatkin and Barton 1989), which is a measure of population differentiation based on genetic polymorphisms.
Population structure
Among-population variability was studied using both distances (1) and frequencies (2) using cluster techniques. Generally, distance methods (1) were based on “mutation” and “non-mutation” models. The distance method (geometric distance) has been recommended for microsatellite data and for populations under the presence of bottleneck effects (Tekezaki and Nei 1996). To avoid confusion, and to interpret data with caution, we opted for the geometric distance (i.e. “non-mutation”) models. The calculations were performed by using R V. 2.5.1. (Ihaka and Gentleman 1996). The confidence values for each clade that was generated by the different clustering techniques were estimated by using permutation and a bootstrapping approach with 103 replications (Felsenstein 1985; Raymond and Rousset 1995a; Holmes 2003). The NJ algorithm for tree building was considered to be preferable to the unweighted pair group method with arithmetic mean (UPGMA) after preliminary tests using TREEFIT ver. 1 software (R 2av = 0.94 for F st; Kalinowski 2009).
In the frequency (2) model-based approaches in population genetics, we utilised the Bayesian methods. Individuals were assigned to populations based on their multilocus genotypes using the model implemented in STRUCTURE V.2.2 (Pritchard and Wen 2003; Falush et al. 2003). The analyses were performed under the “non-admixture” model. Pre-determination of the number of clusters was performed by 50 independent runs for each scenario of the number of true clusters (K; 1–9) in our data under the following conditions: chain length of 106 as a burn-in period, with an additional 106 for the Markov Chain Monte Carlo (MCMC) process. The conserved true clusters were obtained by calculating the maximal values of two coefficients: (a) the second-order rate of change in the likelihood (ΔK) and (b) the logarithm of the mean likelihood L(K), respectively, following Evanno et al. (2005). Generally, both coefficients will show the same result if among-population structure is not too complex. The true cluster must show the maximum constant logarithms of probability using data (Ln P(D)) at each interaction in the period of analysis. Among-population structure that was determined by distance and frequency methods was completed by analysis of molecular variance (AMOVA; Excoffier et al. 1992), with the aim to determine genetic variations among groups of populations and among populations within groups.
The Hardy–Weinberg equilibrium (HWE) was evaluated for the genotypes of 144 and 72 individuals, respectively, from “Cluster-1” and “Cluster-2”. These are two genetically homogeneous regions confirmed from two different cluster technics: distance and frequency (i.e. Roger’s 1972 geometric distance and STRUCTURE V.2.2) algorithms at 104 replications by Fisher’s exact test implemented in the GENEPOP ver. 4 program (Raymond and Rousset 1995b). HWE assumes that evolutionary forces (mutation, migration and natural selection) are negligible. The large sample size was used to avoid cumulative “sampling effects” introduced by sampling error (Hartl and Clark 1997; Hedrick 2000).
Demographic history
The contemporary effective population size (N e) for P. nigra was estimated with summary statistics and an approximate Bayesian computation as described by (Tallmon et al. 2004). The method is based on simulation of a single population. We used lower and upper bounds on the prior for N e of 2 and 1000, respectively. Generally, estimates of N e are typically much lower than census size, so beforehand, we tested upper bounds of 25,000 and 125,000 to help us in the choice of prior to be used with the present data.
The population size fluctuations were studied by two different model-based methods. We used Bayesian (1) coalescent-based approach of Beaumont (1999; MSVAR V.0.4.1.b software). This method assumed a single-step mutation model and estimates the posterior distribution of the rate of population change, i.e. ratio (r) between the current effective number of chromosomes and number of chromosomes at the time of population decline or expansion. For declining populations, log10(r) values are <l, and for population in expansion log10(r) is ≥1. We tested preliminary exponential and linear models and different rectangular priors, but the differences were not significant, and we conducted 10 independent tests replicating MCMC simulations that showed more stability in the rectangular prior (limit of −8 to 8 with the linear model). Each run was conducted with 2 × 109 replications (105 steps by 2 × 104 thinned update). The first 10 % were discarded as burn-in, and the remaining data were used to obtain the mode of 10, 50 and 90 % highest posterior densities (HPD) of the posterior distributions for each parameter, using the module LOCFIT V.2.0 of statistical package R (www.r-project.org).
The second model-based method (2) generated M-statistics, according to Garza and Williamson (2001). M-ratio is the ratio (M = k/r) of the number of observed alleles (k) to the range (r) in allele size for a sample of microsatellite loci, which can be used to detect reductions in effective population size. An equilibrium distribution of Mr was simulated according to the method described in Garza and Williamson (2001), given assumed values for the three parameters of a two-phase mutation model (TPM), and the calculated value is ranked relative to the equilibrium distribution. The critical M-value (Mc) was calculated for 104 replications of each population. The average M-ratio (Mr), less Mc, is an indication of recent reductions in population size (if not biased by a single locus). This method was used as complementary to the first method. The practical difference between them is that the method of Beaumont (1999) covers long periods back in time, while the Garza and Williamson (2001) method is used only for recent population demographic events (i.e. ≤4N e). For example, N e = 100 recent population reduction methods as M-stat of Garza and Williamson (2001) does not reach more of than 400 generations or about 8000 years BP (at reproductive ages of 20 years).
Results
All the analysed nuclear microsatellite loci were polymorphic, and the fragment length size varied between 132 and 325 bp. Four to twelve alleles were found at each locus (Appendix 1). A total of 87 alleles were identified at the 12 loci. At the population level, the expected heterozygosity (H e) was lower in Pop-E and Pop-I, from the Osogovo (W Bulgaria) and St. Planina Mountains (Central Bulgaria), than other provinces included in this study. The average unbiased genetic diversity (U h) was 0.50 (Table 1). Allele richness (A a) and the effective number of alleles (A e) were much lower in the Gabra provenance (Pop-I) from Osogovo Mountains. Private alleles were not observed in the four populations that were dispersed over the range of natural distribution of Pinus nigra in S–W Bulgaria (Pops-C, D, G and I). Two of these populations (Pop-D and I) did not have the local common alleles (<25 %).
Population structure and migration
The neighbour-joining (NJ) geometrical distance shows the presence of three groups. The first two Rhodopes/Slavianka (S–W Bulgaria/“Cluster-1”: Pops-A, C, D, G) and St. Planina/Sr. Gora (Central Bulgaria/“Cluster-2”: Pop-E, F) Mountains are well defined by bootstraps values (>50 %). The third group (S–W Bulgaria/“Cluster-3”) is not well supported by bootstrap values, and each population is from a different mountain; Pop-B from Rila, Pop-H from Pirin and Pop-I from Osogovo Mountains, respectively (Appendix 2).
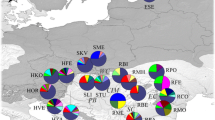
The complementary analysis of Bayesian statistics generally supports the similar structure. Using STRUCTURE V.2.2, the mean likelihood and the second-order change in the likelihood (ΔK) consistently indicated the presence of four clusters in our allelic frequency data (data not shown). The average proportion of membership for each population ranged between 0.331 and 0.525 (F st = 0.223). This is a result of the relatively large effective population size (N e ≈ 686) and very intensive sampling effort in such a relatively small area (about 44,000 km2; Fig. 1). The “non-admixing” cluster makes clades for each principal mountain regions (Fig. 2). “Cluster-1” includes the Mountains Rhodopes and Slavianka (S–W Bulgaria); “Cluster-2” St. Planina and Sr. Gora (Central Bulgaria); “Cluster-3a” Pirin; and “Cluster-3b” Rila and Osogovo (S–W Bulgaria). The significant conformity with HWE (p < 0.004), as shown by Fisher’s exact test, suggested that random mating occurred between individuals in each of the two European black pine formations, i.e. “Cluster-1” and “Cluster-2” (data not shown). AMOVA showed an among-population variability of 13.31 %, which was similar to the G st (12.51 %). AMOVA from hierarchical clustering estimated percentages of variation ranged between 5.58 and 8.01 % among groups of populations, respectively, depending on the method used. Lower among-population variability values within groups were obtained based on the structure produced by the Bayesian algorithm, using stochastic optimisation (data not shown).

Membership of individuals from the nine populations to the four clusters (K = 4) determined by the STRUCTURE V.2.2 model for correlated allele frequencies with “non-admixture”. The bar graph shows the membership to each cluster (each colour corresponds to a different cluster). The analysis was performed by pooling all genotypes without initial population labelling (i.e. non pre-determined number of clusters). The values of F st for each cluster at each interaction and allele-frequency divergence among four clusters. The average F st = 0.223, the average expected heterozygosity H E = 614.46
The number of migrants per generation, N m that was estimated from F st, was 1.63, which indicated the average symmetric gene flow. The P. nigra among-population variability in Bulgaria is highest in that obtained in Asia Minor (Turkey) and lowest from the extreme western part (Spain and Morocco) of its distribution (Cengel et al. 2012; Rubio-Moraga et al. 2012).
Contemporary effective population size and demographic history
Estimates of contemporary effective population size (N e) for the European black pine were 686.0 (median) for the upper prior 1000 and a confidence limit of 95 % (CL 191.1–10,212.1). The obtained average values of N e were therefore used for determining the timing of population decline log10(t f ), from method of Beaumont (1999).
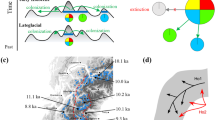
The result of Bayesian coalescent-based approach strongly supports the outcome that the population size declined by 90, 50 and 10 % HPD for log10(r) < 1. The average contemporary population is 10.5 % (at 10 % HPD) of its historical size, suggesting a strong decline in the P. nigra. The observed bottleneck occurred many generations in the past given the posterior distribution for log10(t f ) = 2.40 (at 10 % HPD; Table 2; Fig. 3). The average mode of log10(t f ) indicates that decline had stopped since +2.19 values for all populations studied (at 10 % HPD).

Plots of 54,000 simulated points (18,000 by colour) from the marginal posterior distribution of Log10(r) and Log10(t f ) for Pop-B (red), Pop-A (blue) and Pop-E (green) populations of European black Pine. The solid lines give the 90, 50 and 10 % HPD limits estimated from microsatellite data. Note that the horizontal axes Log10(r) < 1 (and >1) indicate decline (and expansion) in population size, respectively, but the vertical axes Log10(t f ) > −1 are synonymous to the timing of the demographic events back in time. The axis X and axis Y are Log10(r) and Log10(t f ), respectively. The pattern of Pop-G is similar to Pop-B; the pattern of Pop-A (blue) shows four waves of population size decline; remaining populations have similar pattern as Pop-E, i.e. long genealogy, with strong bottleneck (Log10(r) < −1) within very, very old population decline in red/left (Log10(t f ) ≈ 5.5 at 10 % HPD) and old population decline in green/left (Log10(t f ) ≈ 1.8 at 10 % HPD)
The complementary tests through M-stat support the hypothesis of a recent bottleneck. The average M-ratios (0.443) that were calculated for all loci were below the threshold M c (0.68) according to our estimates, and for the critical M (M c) for Black pine, ratios are similar to that obtained by Garza and Williamson (2001; mean = 0.684 and 0.664 for IAM and SMM, respectively. The M-stat shows a bottleneck less 4N e generation BP, i.e. 2744 generations (at N e = 686.0; Table 2).
The results of both methods suggest that the European black pine’s population sizes declined in the very, very old pass due to non-anthropogenic activity (probably due to speciation); after that population sizes were stable and started to decline again in the period of human expansion and activity (less 60,000 years BP/at 25 years per generation) in the studded region.
Discussion
Population structure
Populations among the different mountain ranges had distinctive expected heterozygosity, private alleles and effective numbers of alleles. The distance methods based on NJ geometric distances, respectively, yielded two groups well determined for populations separated by the watershed of Marica River, i.e. Rhodopes/Slavianka (Pops-A, C, D and G) and Sr. Gora/St. Planina (Pops-E and F). The Rhodopes/Slavianka population group was more homogeneous (AMOVA; 4.14 %) than the second “NJ cluster” (Pops-E, F; AMOVA; 16.1 %). The Bayesian frequency method (“non-admixing” cluster) confirmed “NJ cluster” structure and better separated populations between the Pirin and Rila Mountains.
The among-population structure of black pine showed a stronger “mountain effect” than that assessed in the previous chloroplast DNA study (Naydenov et al. 2006). The difference between the present results and cpSSR was probably caused by different migration patterns and the effective radius of dispersal of P. nigra pollen and seeds. The high degree of topographic variability in the region is the cause of contrasting microclimatic conditions, which have established or increased the physiological barrier for mating between pines of different origins (Velkov et al. 1983). Studies over the last 50 years for needles, seeds, cones, bark and crown shape have confirmed the existence of geographic isolation in the study region (Kostov 1974; Mihailov 1993, 1998). The association between genetic structure and geographic distance was not significant (Mantel test; data not shown), which seems to have resulted from: (1) sufficient effective population size and founder numbers following glacial depression; and (2) a high level of adaptation in contemporary areas of natural distribution (Dobrinov et al. 1982, Dobrinov 1983; Alexandrov et al. 1988). Some authors have hypothesised that population differentiation in the Balkan Peninsula is of ancient origin and reflects multiple climatic fluctuations during the Pleistocene (Stefanov 1941/1942, 1943; Fukarek 1958a, b; Mirov 1967). The estimated number of migrants (N m) between populations averaged 1.63, which is less than that determined by allozyme analysis. Gene flow estimates among populations that have been obtained from allozyme analysis have historically illustrated the strong migration capacity of this species. The estimated number of migrants varied between 1.6 and 11.1 individuals and across different European regions (mean = 5.54; Nikolic and Tucic 1983; Scaltsoyiannes et al. 1994a, b; Goncharenko and Silin 1997).
One possible scenario for the black pine history, suggested by our data, is that climatic fluctuations limited migration of this species to individual watersheds within a mountain range. During a cool period, contact between different P. nigra formations that were converging towards valley bottoms was limited by competing vegetation and the heliotropic biology of this species. With climate warming, altitudinal migration in the mountain slopes was easier, as P. nigra is not limited by poor soil conditions. In the study region altitudinal expansion was generally limited by Scots pine (Pinus sylvestris L.), which has a biology similar to black pine but is better adapted to high altitude. Black pine was excluded from low elevations by species that were more adapted to the warming (Fagus sylvatica L., Acer ssp., Fraxinus ssp. and Quercus spp.) and limited at higher altitudes by P. sylvestris. Despite natural areas for black pine being highly fragmented, it can survive in arid locations and under poor soil conditions with little vegetation competition. We have not considered the scenario that very large forest fires created the observed patterns, as both forest fire intensity and frequency are low in this region. The observed structural pattern is probably the consequence of historical events and the autecological characteristics of the species and forest fires, rather than geographic distance per se.
Recently published maritime pine’s large area nDNA analysis shows an intensive migration pattern between three big formations in the period of LGM (10,000–25,000 YBP) across two principal barriers: Strait of Gibraltar and Pyrenees Mountain (Fig. 2 in Naydenov et al. 2014). The principal formations (group of populations) are N–W Africa (Morocco), Iberian Peninsulas (Spain and Portugal) and Continental France. The peaks in the figure indicate that 5–6 times natural area of distribution was not fragmented or less fragmented and had the “contact zones” between principal formations. If we speculate that the “contact zone” was 100 km2 in each region—Gibraltar strata and Pyrenees Mountains—the average inter-formation migration will be 1.3–1.6 individuals for one square kilometre (1 km2) per generation in “contact zone”. We believe that inter-formation migration depended on “contact zone” dimension and was not constant. Probably inter-formation maximum migration rate did not exceed 3–5 individuals per generation, i.e. close to the reported results for among-population migration at present day by Mariette et al. (2001), Ribeiro et al. (2001) and Naydenov et al. (2014). We suggest that similar to P. pinaster, in Bulgaria European black pine’s long gene exchange little affected the local genetic pool. This speculation is supported by the presence of multiple natural barriers like mountains, the Danube River, the Black sea and the Sea of Marmara. We hope to test this hypothesis in the near future.
Demographic history
The detection of recent reductions in population size depends on the biological characteristics of the species, such as lifespan, migration rate and inbreeding tolerance, which are factors that must be considered in both methods that were used. For real endangered populations that have suffered from demographic decline and that have short life spans and long-distance dispersal, including some species of insects, fish and mammals, the discovery of bottlenecks is difficult (Whitehouse and Harley 2001; Jones et al. 2004; Watts et al. 2004). Additional factors have been invoked to control accuracy of bottleneck detection, including patterns of demographic declines and past founder effects (Lippe et al. 2006; Fave and Turgeon 2008). Fragmented populations with small population sizes and low genetic diversity may experience prolonged bottlenecks, which in turn may lead to severe erosion of the gene pool.
The results based on statistically proven methods adapted to nSSR show reductions in population size in P. sylestris, P. halepensis and P. pinaster populations from the Mediterranean region and the south-eastern Europe (Salim et al. 2010; Naydenov et al. 2011, 2014). The comparison with the present study shows that the effective number of alleles (A e ) and the unbiased expected heterozygosity (U h ) have lowest values for Aleppo pine, followed by maritime pine, European black pine and highest for Scots pine. The recent bottleneck (M-ratio) is less pronounced for P. nigra, followed by P. halepensis, P. sylvestris and highest for P. pinaster populations. However, the old bottleneck has lowest impact (Log 10 r) for maritime pine, followed by Scots pine, European black pine and highest for Aleppo pine populations. Furthermore, the timing of the bottleneck is more recent for P. sylvestris, followed by P. pinaster, P. nigra and least recent for P. halepensis populations. The demographic history is different for each species and depends on multiple factors which are not yet well known. We speculate that: (1) for some species rich in multiple subspecies, such as P. nigra, or (2) having closely related species, such as P. halepensis with its close congeners P. brutia Ten. and P. eldarica Medw., they have resulted from great reductions in population size for a very, very long time.
We have used more loci and a larger sample size than previous studies of the same species in order to minimise the risk of size homoplasy. The presence of “null” alleles and homoplasy is possible, but it has been shown that their impact is negligible if the sample size is >30 (Krutovskii et al. 1999; Mariette et al. 2001; Derory et al. 2002).
Conservation and management
Over the past 50 years, the Bulgarian Forest Service has heeded recommendations that seeds of local origin be used for reforestation, based on morphological and physiological analyses (Dobrinov et al. 1982; Dobrinov 1983; Naydenov et al. 2003). This policy was implemented to conserve genetic diversity that had been previously documented in black pine, and our results tend to support the continuation of this strategy. However, the policy should go further and apply local seed collection, extraction and conservation in order to avoid mixing of the seed samples from different provinces, together with a permanent and robust archive system where the origin of seeds used in reforestation should be established. A large number of seed source provenance trees should be maintained, and these individuals must be protected from forest fire and illegal harvesting. Furthermore, long-term planning should include strategies for replacing old seed sources with forest sources from the same genetic pool.
During the last half-century, a significant number of P. nigra trees have been planted outside their natural distribution area in Bulgaria. As past bottlenecks and small founding populations of seed sources can result in excessive inbreeding (Zhelev 1992; Zhelev et al. 1994), questions related to the effective population size of the reproductive individuals should be addressed. Inbreeding in these newly established populations would be deleterious, as it may affect individual reproductive capacity or fitness by increasing the expression of deleterious mutations. In such cases, re-introduction efforts must attempt to reflect the genetic structure of the original population by introducing individuals that are not too divergent. Re-introduction of species through reforestation programmes is vulnerable to outbreeding depression among individuals with widely different genetic makeup. For future generations, the risk of affecting fitness-related traits is high. Depending on the biology and outbreeding level of a particular species, two possible scenarios are possible: (1) local extinction or (2) natural selection towards elimination of ill-adapted genotypes in future generations, with survival of a few more vigorous hybrids. Robust molecular analysis is therefore necessary to compare currently established material with new reforestation projects.
In the last 20 years, economic changes in Eastern Europe have influenced forest practices. The restitution of forestry, in some cases, has been negative for genetic diversity conservation and management. Forest holders in difficult economic situations have harvested important sources of seeds for reforestation, without any control. This phenomenon is more important for the black pine in region because its area of distribution is in a region of restitution, or close to it. This means that the natural area of the black pine is in the area that had been returned to private owners. The second problem is lack of adequate funding for forest protection. Illegal harvesting in public areas, national parks and expansion of tourism over the last 20 years has also had important negative impact. The expert statements confirm that the average annual volume of illegally logged timber for industrial use in Bulgaria in 2003 is about 1.0 million m3. This only includes the uncovered and recorded cases of illegal harvesting. The same experts affirmed that a lion’s share of illegal activities in forests is not recorded (Kostov et al. 2005). The analysis that we have presented here shows the fragility of biodiversity in the region and the need for more adequate programmes in its conservation.
Conclusion
Nuclear DNA analysis of European black pine from sampling sites across Bulgaria showed a moderate level of differentiation among populations. In our study, the proportion of total genetic diversity that was partitioned among populations was 13.31 %, which is a higher level of differentiation than has been previously reported for this species in this region based on allozyme analysis (Naydenov et al. 1993/1996). Gene flow was low (≈1.63), and the presence of a bottleneck was evident. Different clustering methods confirmed the presence of the “mountain effect” in its structure. Conservation/management efforts are urgently required due to intensive anthropogenic pressures and limited natural areas for European black pine in the region.
The international forest tree meta-genomic projects developed in Europe (EU) such as ProCoGen, EvolTree, TreeBreDex and NovelTree reported very good initial results over the last 5–10 years. The coniferous, especially the Genus Pinus’s, members have the biggest genomes in the world, and this has been the principal problem for full genome mapping. The present results for phylogeographic structure of P.nigra are complementary, and they would have been very expensive and difficult to obtain other techniques.
References
Alexandrov A, Rafailov G, Nedelin G, Canov K, Bogdanov B, Spasov C (1988) Coniferous forest in Bulgaria. Zemizdat, Sofia [in Bulgarian]
Auckland LD, Bui T, Zhou Y, Shepherd M, Williams C (2002) Conifer microsatellite handbook. Corporate Press, Raleigh
Beaumont MA (1999) Detecting population expansion and decline using microsattelites. Genetics 153:2013–2029
Belkhir K (2002) GENETIX V. 4.01 A software for population genetics data analysis. Laboratoire Génome et populations, Université de Montpellier II, France
Bondev I (1973) Atlas of Bulgaria. Bulgaria Academy of Sciences, Sofia [in Bulgarian]
Cengel B, Tayanc Y, Kandemir G, Velioglu E, Alan M, Kaya Z (2012) Magnitude and efficiency of genetic diversity captured from seed stands of Pinus nigra (Arnold) subsp. pallasiana in established seed orchards and plantations. New For 43(3):303–317
Derory J, Mariette S, Gonzalez-Martınez SC, Chagne D, Madura D, Gerber S, Brach J, Persyn F, Ribeiro MM, Plomion C (2002) What can nuclear microsatellites tell us about maritime pine genetic resources conservation and provenance certification strategies? Ann For Sci 59:699–708
Dobrinov I (1983) Genetic and selection of forest trees. Zemizdat, Sofia [in Bulgarian]
Dobrinov I, Doykov G, Gagov V (1982) Forest genetic pool in Bulgaria. Zemizdat, Sofia [in Bulgarian]
Evanno G, Madec L, Arnaud JF (2005) Multiple paternity and post-copulatory sexual selection in a hermaphrodite what influences sperm precedence in the garden snail Helix aspersa? Mol Ecol 14:805–812
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Schneider S, Roessli D (2002) ARLEQUIN V.2.001 A software for population genetics data analysis. Department of Anthropology and Ecology, University of Geneva, Geneva
Falush DM, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data linked loci and correlated allele frequencies. Genetics 164:1567–1587
Fave MJ, Turgeon J (2008) Patterns of genetic diversity in Great Lakes bloaters (Coregonus hoyi) with a view to future reintroduction in Lake Ontario. Cons Gen. doi:10.1007/s10592-007-9339-6
Felsenstein J (1985) Confidence limits on phylogenies an approach using the bootstrap. Evolution 39(4):783–791
Fukarek P (1958a) Prilog poznavanju cgnog bora. Radovi, Poljoprivredno-sumarskog fakulteta Univerziteta u Sarajevu 3:3–146 [in Serbian]
Fukarek P (1958b) Die Standortsrassen der Schwarzfohre (Pinus nigra Arn.). Centralbl F d ges Forstwesen 75:203–207 [in German]
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite DNA. Mol Ecol 10:305–318
Goncharenko GG, Silin AE (1997) Populyatsionnaya i evolyutsionnaya genetika sosen Vostochnoi Evropy i Sibiri. [Population and Evolutionary Genetics of Pine in Eastern Europe and Siberia.] Tekhnalogiya, Minsk, Belarus [in Russian]
Hartl DL, Clark AG (1997) Principles of population genetics, 3rd edn. Sinauer Associates Inc., Sunderland
Hedrick P (1996) Bottleneck (s) or metapopulation in cheetahs. Cons Biol 10:897–899
Hedrick PW (2000) Genetics of populations, 2nd edn. Jones and Bartlett, Boston
Holmes S (2003) Bootstrapping phylogenetic trees theory and methods. Stat Sci 18:241–255
Ihaka R, Gentleman R (1996) R A language for data analysis and graphics. J Comput Graph Stat 5:299–314
Jones ME, Paetkau D, Geffen E, Moritz C (2004) Genetic diversity and population structure of Tasmanian devils, the largest marsupial carnivore. Mol Ecol 13:2197–2209
Kalinowski ST (2009) How well do evolutionary trees describe genetic relationships between populations? Heredity 102:506–513
Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17:230–241
Kimura M, Crow JF (1964) The number of alleles that can be maintained in a finite population. Genetics 49:725–738
Kostov K (1974) A very insect-resistant form of Pinus nigra (Arn.) in Bulgaria. For Manag (Bulgaria) 3:6–16 [in Bulgarian]
Kostov G, Paligorov I, Petrov S, Bogdanov Z (2005) Illegal logging in Bulgaria. In: Hirschberger P (ed) WWF European Forest programme and the Danube Carpathian Programme (DCP)
Krutovskii KV, Erofeeva SY, Aagaard JE, Strauss SH (1999) Simulation of effects of dominance on estimates of population genetic diversity and differentiation. J Hered 60:499–502
Leberg PL, Firmin BD (2008) Role of inbreeding depression and purging in captive breeding and restoration programmes. Mol Ecol 17:334–343
Lehman N (1998) Conservation biology genes are not enough. Curr Biol 8:R722–R724
Lian C, Miwa M, Hogetsu T (2000) Isolation and characterization of microsatellite loci from the Japanese red pine, Pinus densiflora. Mol Ecol 9:1171–1193
Lippe C, Dumont P, Bernatchez L (2006) High genetic diversity and no inbreeding in the endangered copper redhorse, Moxostoma hubbsi (Catostomidae, Pisces) the positive sides of a long generation time. Mol Ecol 15:1769–1780
Mariette S, Chagnea D, Lezier C, Pastuszka P, Raffin A, Plomion C, Kremer A (2001) Genetic diversity within and among Pinus pinaster populations: comparison between AFLP and microsatellite markers. Heredity 86:469–479
Mihailov V (1993) Biological and morphological study of European Black pine (Pinus nigra Arn.) seeds from different provenances and selection structures in Pirin and Slavianka mountains in Bulgaria. Ph.D. dissertation, Forest Research Institute, Bulgarian Academy of Sciences, Sofia, Bulgaria [in Bulgarian]
Mihailov V (1998) Variability of European Black pine (Pinus nigra Arn.) according to the size, weight and shape of cone apophyses in Pirin and Slavianka mountains. For Sci (Bulgaria) 1–2:24–37 [in Bulgarian]
Mirov NT (1967) The genus Pinus. Ronald Press, New York
Naydenov KD, Velkov D, Alexandrov A, Genov K, Asparuchova E, Iliev I (1993/1996) Research on chemophenotypic variation of representatives in the genus PINUS with regards to their preservation—Rapport N CC-318/93 NFNI-MONT, Bulgaria [in Bulgarian]
Naydenov KD, Tremblay MF, Ganchev P (2003) Karyotype diversity in of European Black pine (Pinus nigra Arn.) from Bulgarian provenances. Phyton 43(1):9–28
Naydenov KD, Tremblay F, Fenton N, Alexandrov A (2006) Structure of Pinus nigra Arn. populations in Bulgaria revealed by chloroplast microsatellites and terpenes analysis Provenance tests. Biochem Syst Ecol 34:562–574
Naydenov KD, Naydenov MK, Tremblay F, Alexandrov A, Aubin-Fournier LD (2011) Patterns of genetic diversity that result from bottlenecks in Scots Pine and the implications for local genetic conservation and management practices in Bulgaria. New For 42:179–193
Naydenov KD, Alexandrov A, Matevski V, Vasilevski K, Naydenov MK, Gyuleva V, Carcaillet C, Wahid N, Kamary S (2014) Range-wide genetic structure of maritime pine predates the last glacial maximum: evidence from nuclear DNA. Hereditas 151:1–13
Nei M (1975) Molecular population genetics and evolution. North-Holland, Amsterdam
Nikolic D, Tucic N (1983) Isoenzyme variation within and among populations of European Black pine (Pinus nigra Arn.). Silvae Genet 32:80–89
Peakall R, Smouse PE (2006) GENALEX V. 6 genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pertoldi C, Bijlsma R, Loeschcke V (2007) Conservation genetics in a globally changing environment present problems, paradoxes and future challenges. Biodivers Conserv 16:4147–4163
Pimm SL, Gittleman JL, McCracken GF, Gilpin M (1989) Plausible alternatives to bottlenecks to explain reduced genetic diversity. Trends Ecol Evol 4:176–178
Pritchard JK, Wen W (2003) Documentation for structure software version 2 [online]. http://pritch.bsd.uchicago.edu/software/readme_2_1/readme.html. Accessed 17 June 2005
Raymond M, Rousset F (1995a) An exact test for population differentiation. Evolution 49:1280–1283
Raymond M, Rousset F (1995b) GENEPOP: a population genetics software for exact test and ecumenicism. J Hered 86:248–249
Ribeiro MM, Plomion C, Petit RJ, Vendramin GG, Szmidt AE (2001) Variation in chloroplast single-sequence repeats in Portuguese maritime pine (Pinus pinaster Ait.). Theor Appl Genet 102:97–103
Rogers JS (1972) Measures of genetic similarity and genetic distance. Univ Tex Publ 7213:145–153
Rubio-Moraga A, Candel-Perez D, Lucas-Borja ME, Tiscar PA, Viñegla B, Linares JC, Gómez-Gómez L, Ahrazem O (2012) Genetic diversity of Pinus nigra Arn. Populations in Southern Spain and Northern Morocco revealed by inter-simple sequence repeat profiles. Int J Mol Sci 13:5645–5658
Salim K, Naydenov KD, Benyounes H, Tremblay F, Latifa EH, Wahid N, Valeria O (2010) Genetic signals of ancient decline in Aleppo pine populations at the species’ southwestern margins in the Mediterranean Basin. Hereditas 147:165–175
Scaltsoyiannes A, Rohr R, Panetsos K, Tsaktsira M (1994a) Allozyme frequency distributions in five European populations of Black pine (Pinus nigra Arnold). I. Estimation of genetic variation within and among populations. Silvae Genet 43:20–25
Scaltsoyiannes A, Rohr R, Panetsos K, Tsaktsira M (1994b) Allozyme frequency distributions in five European populations of Black pine (Pinus nigra Arnold). II. Contribution of isozyme analysis to the taxonomic status of the species. Silvae Genet 43:25–30
Shannon CE (1948a) A mathematical theory of communication. Bell Syst Tech J 27:379–423
Shannon CE (1948b) A mathematical theory of communication. Bell Syst Tech J 27:623–656
Slatkin M (1987) Gene flow and geographic structure of natural populations. Science (Washington, D.C.) 236:787–792
Slatkin M, Barton N (1989) A comparison of three indirect methods for estimating average levels of gene flow. Evolution 43:1349–1368
Stefanov B (1941/1942) Geographical distribution of coniferous species and their form in nature. Godichnik na Sofiiskia Darjaven Universitet, Sofia, (Bulgaria), XIX and XX, 1–88 [in Bulgarian]
Stefanov B (1943) The phyto-geographical elements of Bulgaria. Thesis of Bulgarian Academy of Sciences, Faculty of Nature and Mathematics, Sofia, (Bulgaria), Vol. XXXIX, 19:1–121 [in Bulgarian]
Tallmon DA, Beaumont MA, Luikart GH (2004) Effective population size estimation using approximate Bayesian computation. Genetics 167:977–988
Tekezaki N, Nei M (1996) Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 144:389–399
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) Program note: MICRO—CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Velkov D, Mihailov V, Dobrev R (1983) Taxonomical and biological study of seed production practices for Pinus nigra (Arn.). In Nauchno-technicheska konferencia s mejdunarodno uchastie na tema “Nasoki I problemi na izgrajdaneto na gorskata semeproizvoditelna baza”, Borovec, Bulgaria. Vol 1, pp 12–13 [in Bulgarian]
Vidakovic M (1974) Genetics of European Black pine (Pinus nigra Arn.). Ann For (Zagreb) 6:57–86
Vidakovic M (1991) Conifers—morphology and variation. Graficki Zavod Hrvatske, Croitia
Watts BD, Byrd MA, Watts MU (2004) Status and distribution of breeding Ospreys in the Chesapeake Bay 1995–1996. J Raptor Res 38:47–54
Whitehouse AM, Harley EH (2001) Post-bottleneck genetic diversity of elephant populations in South Africa, revealed using microsatellite analysis. Mol Ecol 10:2139–2149
Willi Y, Van Buskirk J, Hoffmann AA (2006) Limits to the adaptive potential of small populations. Annu Rev Ecol Evol Syst 37:433–458
Williams CG, Elsik CG, Barnes RD (2000) Microsatellite analysis of Pinus taeda L. in Zimbabwe. Heredity 84:261–268
Wrigth S (1965) The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 19:395–420
Zhelev P (1992) Ecolo-biological and selection—genetic Scots Pine investigations from the Rhodope Mountains. Ph.D. Thesis of forest genetic, selection and tree breeding. University of Forestry, Sofia, Bulgaria [in Bulgarian]
Zhelev P, Longauer R, Ladislav P, Gomory D (1994) Genetic variation of the indigenous Scots Pine (Pinus sylvestris L.) populations from the Rhodope Mountains. For Sci (Bulgaria) 3:68–76 [in Bulgarian]
Acknowledgments
We thank Irena M. Naydenova and T&T for their technical assistance. We are grateful to the staff of the Bulgarian forest service for their assistance with the plant material; the Ministry of Education and Science of Bulgaria (EU) for their financial support (CC-318/1993–1996); and the Fond KAZAROVI-Geneva (Switzerland) for a student grant to K. D. Naydenov during an internship at the University of Geneva, Switzerland (Dept. of Botany). We also wish to thank Dan MacKay (CFLS-St-Jean, Qc., Canada) and Translation-Group (UK) for his careful revision of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Jarmo Holopainen.
This article is dedicated to the memory of Prof. Dr. Kosta Kostov from Forest Research Institute, Bulgarian Academy of Science (1924–2009).
Appendices
Appendix 1
See Table 3.
Appendix 2
The neighbour-joining (NJ) dendrogram of Rogers’s (1972) genetic distance assumes the “non-mutation” model (i.e. geometric distance). The ring on the nodes indicates bootstrap values of ≥50 % at 1000 replications (R 2av = 0.94 for F st; Kalinowski 2009).

Rights and permissions
About this article

Cite this article
Naydenov, K.D., Mladenov, I., Alexandrov, A. et al. Patterns of genetic diversity resulting from bottlenecks in European black pine, with implications on local genetic conservation and management practices in Bulgaria. Eur J Forest Res 134, 669–681 (2015). https://doi.org/10.1007/s10342-015-0881-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10342-015-0881-3