
Abstract
A simple, rapid, isocratic, and versatile liquid chromatographic method was developed for the simultaneous determination of bromhexine, guaifenesin, ambroxol, salbutamol/terbutaline, pseudoephedrine, triprolidine, and chlorpheniramine maleate in cough–cold syrups commonly marketed in Kenya. Separation was achieved using a Gemini® NX C18 column (250 × 4.6 mm, 5 μm) maintained at 40 °C and a mobile phase consisting of acetonitrile-0.25 M sodium hexanesulphonate-0.2 M ammonium acetate, and pH 3.0-water (35:4:10:51, % v/v/v/v) delivered at 1.0 mL min−1. The eluents were monitored by means of UV detection at 254 nm. During validation, the method satisfied the International Committee on Harmonization acceptance criteria for linearity, sensitivity, precision, accuracy, and robustness. The developed liquid chromatographic method was applied in the analysis of nine commercial samples obtained from Nairobi City County, Kenya. Extraction procedures were not applied during the assay of the samples, thus significantly shortening the analysis time.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cough is a non-specific symptom of respiratory disease with broad differential diagnosis. Acute cough may be caused by the common cold, acute sinusitis, pertussis, chronic obstructive pulmonary disease (COPD) exacerbations, and allergic or non-allergic rhinitis. Dry coughs are managed with antitussives, such as codeine, dextromethorphan, and levorphanol, while treatment of productive cough involves use of antibiotics and various combinations of antihistamines, decongestants, mucolytics, expectorants, and bronchodilators [1–4].
Most cough syrups in the Kenyan market contain the study drugs, as several combinations composed of 2–4 bronchodilator-mucolytic-expectorant-antihistamine mixtures. However, some products contain bromhexine or ambroxol as single entities used in the management of COPD. Cough syrups are not listed in the Kenya Essential Medicines List [5]. Furthermore, most hospitals do not include them in their formularies, but medical practitioners prescribe them for patients who, in turn, purchase them from retail pharmacies [6].
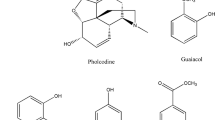
The title drugs are classified as bronchodilators (salbutamol, terbutaline), mucolytics (bromhexine, ambroxol), expectorants (guaifenesin), decongestants (pseudoephedrine), and antihistamines (triprolidine, chlorpheniramine) [1]. These compounds possess varying structural and spectral features that render the development of quality control methods for simultaneous analysis difficult (Fig. 1).

Chemical structures of the compounds under study
High-performance liquid chromatography (HPLC) is the method of choice for the analysis of multi-component products, including cough–cold syrups, due to its superior selectivity and specificity. Reversed-phase HPLC of basic compounds is usually associated with poor selectivity and peak tailing [7]. All the compounds under study are basic except guaifenesin. Excipients in cough-cold products bring about additional analytical challenges.
The British Pharmacopoeia (BP) and United States Pharmacopeia (USP) specify titrimetry or HPLC for the assay of bromhexine (BXN), guaifenesin (GFN), ambroxol (AMB), salbutamol (SBT), terbutaline (TBT), pseudoephedrine (PED), triprolidine (TBN), and chlorpheniramine (CPM) bulk samples, while the corresponding single component products are analyzed by HPLC and UV spectroscopy [8, 9]. The determination of these compounds in admixture with other drugs using reversed-phase HPLC has been reported by various authors. Some of these methods apply gradient elution with long run times given the required equilibration interval [10, 11]. The isocratic methods reported involve the separation of 2–3 of the compounds and, therefore, not applicable for a wide range of actives [12–15]. So far, no HPLC method has been described for the simultaneous determination of all eight compounds in cough–cold remedies. Such a method could be widely applied in the analysis of cough–cold syrups-containing any combination of the named compounds. This paper reports on the development and validation of a simple, rapid, isocratic, and versatile method for the determination of bromhexine, guaifenesin, ambroxol, salbutamol/terbutaline, pseudoephedrine, triprolidine, and chlorpheniramine in cough–cold syrups. The method does not require sample pre-treatment which greatly reduces analysis time and saves on reagents.
Experimental
Chemicals
Analytical grade KH2PO4, K2HPO4, sodium butanesulphonate acid sodium (Loba chemie PVT LTD, Mumbai, India), anhydrous sodium acetate, ammonium acetate, sodium hexanesulphonate, sodium pentanesulphonate, acid sodium, pentanesulfonic acid sodium, triethylamine (RFCL LTD, New Delhi, India), orthophosphoric acid (Merck Chemicals PTY Ltd., Gauteng, South Africa), and heptanesulfonic acid sodium heptanesulphonate (Fischer Scientific, Leicestershire, UK) were used during the experiments.
Acetonitrile (HPLC grade) and glacial acetic acid AR were from Scharlau Chemie S.A. (Sentmenat, Barcelona, Spain). Purified water was prepared in the laboratory by reverse osmosis, ultrafiltration, and UV irradiation using an Arium 61316 RO and Arium 611 VF water system (Sartorius AG, Göttingen, Germany).
Instrumentation
A Shimadzu Prominence HPLC system (Shimadzu Corp., Kyoto, Japan) consisting of a LC-20AT Prominence solvent delivery system, SIL-10AS Prominence autosampler, and SPD-20A Prominence UV/Visible detector was used for the study. The system was supported by a CBM-20A Prominence communications system controller and LC Solutions Software Version 1.22, SP1. The temperature was controlled using a CTO–10AS VP column oven with a block heating thermostatic chamber and a preheater system. Mobile phases were delivered isocratically at a flow rate of 1.0 mL min−1. A Gemini® NX C18 column of dimensions 250 × 4.6 mm, 5 μm (Phenomenex Inc, Torrance, CA, USA) with hybrid RP packing was used for all experiments.
Mobile Phases
Buffer solutions (0.2 M) were prepared by mixing equimolar salt or parent acid solutions to the desired pH. Mobile phases were made by diluting the buffer solutions with water before topping up with acetonitrile. Where ion-pairing agents were employed, stock solutions containing 0.25 M of ion-pairing agent were prepared and added to the buffer solutions before adjusting the pH. Mobile phases were degassed using a power sonic 410 bench top ultrasonic bath (Daihan Labtech Ltd, Kyonggi-Do, Korea).
Working Reference Standards
Working standards of ambroxol hydrochloride (99.4 % w/w), bromhexine hydrochloride (99.8 % w/w), chlorpheniramine maleate (99.9 % w/w), guaifenesin (99.6 % w/w), pseudoephedrine hydrochloride (100.8 % w/w), salbutamol sulphate (99.8 % w/w), terbutaline sulphate (99.7 % w/w), and triprolidine hydrochloride (99.1 % w/w) were a kind donation by the National Quality Control Laboratory (Nairobi, Kenya). The working standards were assayed for potency against USP chemical reference substances before use.
Working Standard Solution
The working standard solution was prepared by dissolving the analytes in acetonitrile-water (40:60 % v/v) to concentrations of AMB 0.04 mg, BXN 0.08 mg, CPM 0.2 mg, GFN 0.5 mg, PED 0.3 mg, SBT 0.3 mg, TBT 0.3 mg, and TPN 0.04 mg mL−1. The solutions were freshly prepared prior to use. The concentrations were designed to yield comparable peak heights for easy derivation of chromatographic parameters. Since SBT and TBT are not co-formulated in drug products, they were incorporated into the reference working solution separately as required.
Method Validation
Validation Standard Solution
The standard solution used for the validation of the method was prepared to the relative concentrations of the analytes similar to that in the commercial products, AMB 0.3 mg, BXN 0.08 mg, CPM 0.02 mg, GFN 0.5 mg, PED 0.3 mg, SBT 0.02 mg, TBT 0.025 mg, and TPN 0.0125 mg mL−1.
Linearity and Range
The validation standard solutions were run at 25, 50, 80, 100, 120, and 150 %. The peak areas obtained were subjected to the linear regression analysis on MS Excel spreadsheets (Microsoft Corporation, Redmond WA, USA).
Precision
Repeatability was determined by making six injections of the validation standard solution on the same day. The peak areas of the analytes were normalized and the coefficient of variation (CV) of the corrected areas computed. For intermediate precision, six replicate injections of a freshly prepared validation standard solution were run on three consecutive days using fresh mobile phases. The peak areas obtained were normalized and the CV thereof used to evaluate the inter-day precision of the method.
Sensitivity
The limit of detection (LOD) and limit of quantitation (LOQ) were determined by preparing serial dilutions of each analyte from stock solutions of concentration equivalent to that of validation standard solution. The signal-to-noise ratio (S/N) values of the individual peaks were determined with reference to the diluent as blank. The LOD was derived from the lowest concentration of the analytes that yielded S/N of 3:1, while S/N 10:1 and peak area CV of 10–20 % were used to establish LOQ [16–18].
Robustness
The influence of the chromatographic factors, pH, temperature, and acetonitrile on separation was tested at three levels, low (−1), central (0), and high (1), as shown in Table 1. The validation standard solution was run after adjusting a factor level and the capacity factors (k′) of component peaks determined. The k′ was plotted against each factor series for the assessment of selectivity.
Accuracy
The accuracy for the developed method was determined by spiking commercial products with working standards of the analytes and determining the recovery upon analysis. Accordingly, different samples were used, since no single product contained all the eight compounds. Recovery was tested at the 80, 100, and 120 % levels. The percentage recovery of each added working standard was used as a measure of accuracy according to the ICH guidance [16–18].
Application
Test Samples
Test samples were purchased from randomly selected retail pharmacies located within the Central Business District and suburbs of Nairobi City County. A total of nine commercial samples were obtained and coded A—I, as shown in Table 2. Six products had three batches analyzed, while two batches of the remaining three products were analyzed due to limited availability.
Sample Preparation
Samples were prepared to concentrations of AMB 0.3 mg, BXN 0.08 mg, CPM 0.02 mg, GFN 1 mg, PED 0.3 mg, SBT 0.02 mg, TBT 0.025 mg, and TPN 0.0125 mg mL−1 in acetonitrile–water (40:60 % v/v). However, there was variation for samples B, C, and G in agreement with the label claims. Standard solutions for quantitation were prepared in tandem.
During sample preparation, 5.0 or 10.0 mL were pipetted into a 100.0 mL volumetric flask and diluted to volume with acetonitrile–water (40:60 % v/v) and filtered prior to the LC analysis. The peak areas of the standard and sample were normalized and used for the quantitation of the active ingredients, whose content was subsequently expressed as percentage of the label claim.
Results and Discussion
Chromatographic Optimization
The detection wavelength (254 nm) was established through overlay of the UV spectra of the individual analytes, while the Gemini® NX column C18 was chosen on account of its pH stability and superior chromatographic parameters for basic compounds according to the previous reports [19]. Acetonitrile was chosen due to its strong eluting power which was desirable as demonstrated in preliminary observations.
Potassium phosphate, sodium acetate, and ammonium acetate buffers pH 3.0 were used with 40 % v/v acetonitrile, whereby 0.02 M ammonium acetate yielded better chromatographic parameters and shorter run time. However, poor selectivity was recorded for SBT/PED/AMB group of peaks. Consequently, ion-pairing agents of the series butane–heptane sulphonate were investigated as means of improving separation, whereof 10 mM hexanesulphonate yielded the best results. Further investigations into the influence of acetonitrile concentration, buffer pH, and temperature yielded the optimum chromatographic conditions as: a mobile phase consisting of acetonitrile-0.25 M sodium hexanesulphonate-0.2 M CH3COONH4, and pH 3.0-water (35:4:10:51, % v/v/v/v) delivered at a flow rate of 1.0 mL min−1 on Phenomenex Gemini® NX column C18, 5 µm, pore size 110 Å of dimensions 250 × 4.6 mm internal diameter maintained at 40 °C. Figure 2 is a typical chromatogram obtained under optimum conditions. Due to structural congruity of SBT and TBT, separation could not be achieved. Therefore, SBT has been used in the chromatogram displays and robustness data for consistency.

Typical chromatogram of the reference working solution obtained under the optimized chromatographic conditions. Column: Phenomenex Gemini® NX C18, 5μ, 250 × 4.6 mm ID. Temperature: 40 °C. Mobile phase: acetonitrile-0.25 M sodium hexanesulphonate-0.2 M CH3COONH4, and pH 3.0–water (35:4:10:51, % v/v/v/v). MAL maleic acid, SBT salbutamol, PED pseudoephedrine, GFN guaifenesin, AMB ambroxol, CPM chlorpheniramine, TPN triprolidine, BXN bromhexine
Method Validation
The validation results obtained are summarized in Table 3. All the analytes demonstrated good linear relationship between concentration and peak area within the range 25–150 % with coefficient of determination, r 2 < 0.999. The LOD and LOQ values indicate the method is sensitive enough for the determination of the analytes in trace amounts. With respect to precision, the repeatability and inter-day variation of the peak areas satisfied the ICH acceptance criteria. The average percentage recovery values of the compounds were within the range 99.2–100.5 % with CV ≤ 1.1, thus indicating adequate accuracy of the method. Samples were screened for any interference of the analyte peaks by the excipients from the sample matrix whereof none was recorded.
Figure 3 shows the effect of pH, temperature, and acetonitrile concentration on the k’ of the peaks. Acetonitrile concentration and pH had the greatest impact on capacity factors, especially the BXN peak. The retention time of BXN was 14.5 min at 37 % v/v acetonitrile and 22.3 min at 33 % v/v. This calls for accurate measurement of mobile phase components and pH to minimize deviations during the application of the method. The resolution of the peaks was >2.0 over the pH, acetonitrile, and temperature ranges tested. The data obtained showed adequate robustness of the method for routine analytical applications.

Effect of variation of the factors on capacity factors of the analytes. SBT salbutamol, PED pseudoephedrine, GFN guaifenesin, AMB ambroxol, CPM chlorpheniramine, TPN triprolidine, BXN bromhexine
Application
Sample Preparation
There was no interference of excipients thus baseline separation was achieved for all the analytes hence no need for extraction prior to HPLC analysis. However, maleic acid co-eluted with some excipients and solvent front, although this was inconsequential, since it was not under determination.
Analysis of Samples
The assay results obtained are summarized in Table 4. For purposes of interpretation, the assay limits of 90.0–110.0 % were adopted from the USP (2015) monographs for finished products. All the samples complied with the specifications for assay. The results obtained also demonstrated that there were minimal batch-to-batch variations.
Conclusion
The developed method is sufficiently versatile for the assay of the eight compounds individually or in combination. The method satisfied the ICH acceptance criteria for selectivity, linearity, robustness, and recovery. It can, therefore, be applied for the routine analysis of cough–cold syrups containing the specified analytes for purposes of market authorization and batch release. The method offers great flexibility during the routine analysis of products containing mixtures of the analytes during routine analysis. The assay results for the products analyzed showed content compliance and batch consistency which is an indicator of good GMP standards maintained by the manufacturers.
References
East African Pharmaceutical Loci (2015) A regional drug index for healthcare practitioners, 14th edn. Pharmaceutical Loci Publishers, Nairobi, pp 244–250
De Blasio F, Virchow JC, Polverino M, Zanasi A, Behrakis PK, Kilinç G, Balsamo R, De Danieli G, Lanata L (2011) Cough management: a practical approach. Cough 7:7
Katzung BG (2010) Basic and clinical pharmacology, 12th edn. McGraw-Hill, New York, p 563
Jongste JC, Shields MD (2003) Chronic cough in children. Thorax 58:998–1003
Ministry of Health (2016) Kenya Essential Medicines List 2016, Nairobi, p 63
Kigen G, Busakhala N, Ogaro F, Chesire E, Saat N, Too R, Nyandiko WA. Review of the ingredients contained in Over the Counter (OTC) cough syrup formulations in Kenya. Are they harmful to infants? PLoS One. 2015; 10(11)
Vervoort RJM, Ruyter E, Debets AJJ, Claessens HA, Cramers CA, de Jong JG (2001) Characterization of reversed-phase liquid chromatography stationary phases for the analysis of basic pharmaceuticals: eluent properties and comparison empirical test methods. J Chromatgr A 931:67–79
British Pharmacopoeia Commission (2015) British Pharmacopoeia. The Stationery Office, London
United States Pharmacopeial Convention ( 2015) United States Pharmacopeia 38 National Formulary 33. United States Pharmacopeial Convention Inc., Rockville, MD, Maryland
Porel A, Haty S, Kundu A (2011) Stability-indicating HPLC method for simultaneous determination of terbutaline sulphate, bromhexine hydrochloride and guaifenesin. Indian J Pharm Sci 73(1):46–56
Mallu UR, Bobbarala V, Penumajji S (2011) Analysis of cough and analgesic range of pharmaceutical active ingredients using RP-HPLC method. Int J Pharm Bio Sci 2(3):439–452
Jain V, Sharma MC (2016) Validated RP-HPLC method for determining the levels of bromhexine HCl, chlorpheniramine maleate, dextromethorphan HBr and guaiphenesin in their pharmaceutical dosage forms. J Taibah Univ Sci 10:38–45
Manassra A, Khamis M, el-Dakiky M, Abdel-Qader Z, Al–Rimawi F (2010) Simultaneous HPLC analysis of pseudoephedrine hydrochloride, codeine phosphate and triprolidine hydrochloride in liquid dosage forms. J Pharm Biomed Anal 51:991–993
Pai PNS, Rao GK, Murthy MS, Agarwal A, Puranik S (2009) Simultaneous determination of salbutamol sulphate and bromhexine hydrochloride in tablets by reversed phase liquid chromatography. Indian J Pharm Sci 71:53–55
Bhatia NM, Ganbavale SK, Bhatia MS, More HN, Kokil SU (2008) RP–HPLC and spectrophotometric estimation of ambroxol hydrochloride and cetirizine hydrochloride in combined dosage form. Indian J Pharm Sci 70:603–608
ICH harmonised tripartite guideline (Q2B) (R1) (2005) validation of analytical procedures: text and methodology. http://www.ich.org
Crowther JB (2001) Validation of pharmaceutical test methods. Sep Sci Technol 3:415–443
Shabir GA (2003) Validation of high-performance liquid chromatography methods for pharmaceutical analysis: understanding the differences and similarities between validation requirements of the US Food and Drug Administration, the US Pharmacopeia and the International Conference on Harmonization. J Chromatogr A 987(1):57–66
Law B, Houghton SJ, Ballard P (1998) An approach to the evaluation and comparison of reversed-phase high-performance liquid chromatography stationary phases. J Pharm Biomed Anal 17:443–453
Acknowledgments
The work presented in this paper was part of Master of Pharmacy thesis of PNM, University of Nairobi (2011). The authors wish to thank the University of Nairobi for financial support and the Management of the National Quality Control Laboratory for providing the necessary equipment to perform this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors of this manuscript have no financial and personal relationships with other people or organizations that could influence their work.
Rights and permissions
About this article

Cite this article
Njaria, P.M., Abuga, K.O., Kamau, F.N. et al. A Versatile HPLC Method for the Simultaneous Determination of Bromhexine, Guaifenesin, Ambroxol, Salbutamol/Terbutaline, Pseudoephedrine, Triprolidine, and Chlorpheniramine Maleate in Cough–Cold Syrups. Chromatographia 79, 1507–1514 (2016). https://doi.org/10.1007/s10337-016-3158-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-016-3158-1