
Abstract
A liquid phase microextraction method using hollow fiber to support extraction solvent was developed for enrichment of trace level chloroanilines in environmental water samples. Target analytes, 2-chloroaniline, 3-chloroaniline, 2,3-dichloroaniline, 2,4-dichloroaniline, 3,4-dichloroaniline, and 3,5-dichloroaniline were determined using gas chromatography-flame ionization detector after extraction. Experimental conditions that affect extraction efficiency were investigated and optimized. The proposed method showed a wide linear range from lower μg L−1 to 1,000 μg L−1, low detection limits (≤5.1 μg L−1), and reasonable relative standard deviations (RSDs < 13%). Feasibility of the method was evaluated by analyzing river water samples collected from the Hudson River and the East River in New York City.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chloroanilines (CAs) are compounds of significant environmental and public health concern because of their toxicity to natural environments and humans [1–3]. They are released into the environment as chemical residues of dyestuff, pharmaceutical, and rubber manufacturing, and as by-products of coal-conversion waste processing [4–6]. CAs are also found as degradation products and intermediates of various phenylurea and phenylcarbamate pesticides [1, 4, 6].
Liquid–liquid extraction (LLE) is a sample preparation technique with a long history, and it is still employed by many laboratories today. However, there are significant disadvantages associated with conventional LLE. For example, the procedure uses relatively large amounts of highly purified organic solvents, and the operation is tedious and time consuming. To avoid these problems, miniaturized liquid-based extraction, termed liquid phase microextraction (LPME), has been developed in recent years. The improved method uses only microliter levels of solvent and integrates analyte extraction and enrichment into a single step. It also features a simple experimental setup and easy combination with common analytical techniques such as gas chromatography (GC) [7–10], high performance liquid chromatography (HPLC) [11–13] and capillary electrophoresis (CE) [14–16]. A high enrichment factor, and consequent low detection limit, can usually be obtained due to the increased volume ratio between sample solution and extraction solvent [17].
Originally, LPME used a solvent drop suspended on the tip of a microsyringe. Since this hanging drop is easily dislodged during extraction, it can be stabilized by attaching a small segment of a hollow fiber to the tip. This is designated two-phase hollow fiber-based LPME. Solvent is impregnated in the lumen and micropores of the hollow fiber. The enlarged solvent surface area and compatibility with higher stirring speeds increases the rate of mass transfer of analytes from sample solution to solvent [17]. The hollow fiber also provides additional filtration to prevent particles from entering the lumen of the fiber. Hollow fiber-based LPME has been shown to be a simple, fast, and inexpensive sample preparation procedure, and has been used to analyze environmental pollutants, biochemical warfare agents, and pharmaceutical residues [9, 18–22].
Solvent drop-based microextraction and solid phase microextraction (SPME) have been reported for the determination of CAs, but generally limited to only one or two CAs [19, 23, 24]. The objective of present study was to develop a hollow fiber-based LPME procedure combined with gas chromatography-flame ionization detector (GC-FID) to monitor trace levels of 6 CAs in environmental water samples. The target analytes were 2-chloroaniline (2-CA), 3-chloroaniline (3-CA), 2,3-dichloroaniline (2,3-DCA), 2,4-dichloroaniline (2,4-DCA), 3,4-dichloroaniline (3,4-DCA), and 3,5-dichloroaniline (3,5-DCA). Experimental conditions affecting extraction efficiency were investigated and optimized. Quantitative performance of the method was evaluated, and method feasibility was tested by analyzing river water samples collected in New York City.
Experimental
Reagents and Standards
Standards of 2-CA, 3-CA, 2,3-DCA, 2,4-DCA, 3,4-DCA, and 3,5-DCA with purities >99% were obtained from Acros (Fair Lawn, NJ, USA). Acetonitrile (Pharmco, CT, USA) was used to prepare 1,000 mg L−1 stock solutions. Q3/2 Accurel polypropylene hollow fibers with 600 μm inner diameter, 200 μm wall thickness, and 0.64 μm pore size were obtained from Membrana (Wuppertal, Germany). Benzene, cyclohexane, n-hexane, octane, octanol, toluene, and sodium hydroxide were purchased from Fisher Scientific (Fairlawn, NJ, USA). Ultra pure water was prepared using a Millipore water filtration system (Bedford, MA, USA).
Hollow fibers were cleaned with acetone and ultrapure water in a sonicator. The dried fibers were cut into 2.5-cm-long segments. After one end of the segment was thermally sealed, it can be used for extraction.
Hollow Fiber-Based LPME
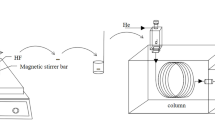
The schematic diagram of a hollow fiber-based LPME setup is illustrated in Fig. 1. Six milliliter of sample solution was transferred to a glass sample vial (8 mL), which was covered with aluminum foil to prevent evaporation. This solution was modified with NaOH to suppress the ionization of the amines. Fifteen microliter of organic solvent was drawn up into a 50-μL Hamilton HPLC syringe (Reno, NV, USA), and a hollow fiber segment was attached to the tip of the syringe. The entire hollow fiber was immersed in the sample solution and the syringe was fastened by a clamp to the iron stand. Solvent was carefully injected into the hollow fiber to start the extraction. The amount injected was determined before extraction. The sample was stirred by a micro-stirring bar through the entire experiment. When extraction was completed, the solvent was slowly withdrawn back into the syringe and the hollow fiber was removed. Usually about 10–12 μL of solvent was recovered, which was transferred to a 200 μL glass insert of a micro-sample vial. 1 μL was taken by a GC syringe for analysis.

Schematic diagram of hollow fiber-based LPME
Instrumentation
A Varian CP-3800 GC-FID (Palo Alto, CA, USA) with a DB-5 column (30 m × 0.320 mm × 1.0 μm, Agilent, Santa Clara, CA, USA) was used for the analysis. The carrier gas was nitrogen at 2 mL/min. The detector flow rates were 300 mL min−1 air, 28 mL min−1 nitrogen makeup, and 30 mL min−1 hydrogen. Temperature of both the injector and detector was 250 °C. A 1:3 split ratio was used for injection. The oven temperature program was 100 °C for 3 min, then ramped at 50 °C min−1 to 210 °C, and held at 260 °C for 5 min.
Environmental Water Samples
Environmental water samples were collected from the East River and the Hudson River in New York. The water samples were stored in a 1 L Nalgene bottle at 4 °C and filtered through 0.2 μm syringe filters (Fisher, Fairlawn, NJ, USA) before use. The samples were analyzed within 3 days after sampling.
Results and Discussion
Optimization of the Experimental Conditions for Hollow Fiber-Based LPME
Optimization of the procedure was based on the one-factor-at-a-time approach, in which the effect of one parameter is investigated and all other parameters are kept constant [25].
Selection of Organic Solvent
Selection of an organic solvent is the first and most important step to establish a LLME procedure. In order to achieve satisfactory analyte enrichment, three factors were considered: (1) the solvent is immiscible with water; (2) it should provide the best overall enrichment for the target compounds; and (3) it should not co-elute with the analytes during GC analysis. Six solvents, namely, benzene, cyclohexane, n-hexane, octane, octanol and toluene were investigated and their extraction efficiencies evaluated by peak areas were compared (Fig. 2). Although octanol exhibited the highest peak response to 2-CA, 3-CA, 2,3-DCA and 2,4-DCA, its extraction to 3,4-DCA and 3,5-DCA were unsatisfactory. Since toluene offered the highest or second highest peak response to all analytes and exhibited good reproducibility, it was chosen as the extraction solvent.

Effect of solvent on extraction efficiency. Extraction conditions: 6 mL sample with 1 M NaOH, no salt addition, 400 rpm stirring speed and 20 min extraction time. The concentration of each CA was 500 μg L−1. Data was based on duplicate analysis
Stirring Speed
Agitation plays an important role in LPME because it effectively enhances mass transfer of target analytes from the sample solution to the solvent phase. Compared with hanging drop LPME, hollow fiber-based LPME significantly improves the stability of the solvent phase so that a higher agitation speed can be employed. Stirring speeds ranging from 400 to 1,200 rpm were investigated. Figure 3 shows that the peak area of each compound increased steadily with the increase of stirring speed. The solvent phase was not dislodged even at 1,200 rpm, the strongest agitation the stirring plate could offer. Stirring speed of 1,200 rpm was thus used in subsequent experiments.

Effect of stirring speed on extraction efficiency. Extraction conditions: 6 mL sample with 1 M NaOH, no salt addition and 10 min extraction time. The concentration of each CA was 500 μg L−1. Data was based on duplicate analysis
Concentration of NaOH in Sample Solution
Because the target analytes were chlorinated aromatic amines, NaOH was added to the aqueous solution to ensure the analytes were in their undissociated forms. NaOH concentrations in the sample solutions were varied from 0.01 to 1 M. The peak area obtained at different concentration levels did not vary significantly (Fig. 4). However, because 1 M NaOH solution provided the best overall extraction results, it was used in subsequent experiments.

Effect of NaOH concentration in sample solution on extraction efficiency. Extraction conditions: 6 mL sample with various NaOH concentration, no salt addition, 1,200 rpm stirring speed and 10 min extraction time. The concentration of each CA was 500 μg L−1. Data was based on duplicate analysis
Effect of Addition of Salt
Addition of salt may either improve the extraction efficiency by decreasing of the aqueous solubility of the organic analytes, or reduce extraction efficiency of polar molecules because of electrostatic interactions with the salt ions [26, 27]. The effect of salt has therefore to be experimentally investigated during method development. The extraction efficiencies for solutions with NaCl concentrations of 0, 10 and 35% were compared and a decreased trend was found for all compounds with the increase of salt content in sample matrix. Because of this reason, no salt was added into sample solution in subsequent work.
Determination of the Extraction Time
The final parameter to investigate was the extraction time. As shown in Fig. 5, the peak area for all analytes increases quickly up to 20 min. 2-CA and 3-CA reach equilibrium at 20 min; however, the peak area of 2,3-DCA, 2,4-DCA, and 3,4-DCA, and 3,5-DCA keeps increasing, but in a slower rate, up to 50 min. Significant solvent loss was observed for extraction time longer than 50 min, which resulted in decreased peak area. In order to balance the extraction efficiency and analysis time, 20 min was chosen as the optimum time in this work.

Effect of time on extraction efficiency. Extraction conditions: 6 mL sample with 1 M NaOH, no salt addition, 1,200 rpm stirring speed. The concentration of each CA was 500 μg L−1. Data was based on duplicate analysis
Quantitative Analysis
The linear range, precision, limits of detection (LODs) and limits of quantification (LOQs) of the hollow fiber-based LPME were investigated under the optimum conditions, i.e. 6 mL sample with 1 M NaOH, no salt addition, 1,200 rpm stirring speed and 20 min extraction time. The results are summarized in Table 1. Good linearity was observed over a wide range from lower μg L−1 to 1,000 μg L−1 for all analytes with R 2 equal or better than 0.9940. The LODs calculated using 3σ/calibration slope [28] were in lower μg L−1 level. The LOQs were three times the LODs. Method precision was evaluated by extracting 100 μg L−1 sample solutions; the relative standard deviations (RSD%, n = 6) of each analyte was less than 13%. The enrichment factors ranging from 81 for 3-CA to 151 for 2,4-DCA were obtained. The proposed hollow fiber-based LPME was comparable to the methods reported in previous studies [19, 29] listed in Table 2, which summarizes the main analytical performances of these methods for determination of 3,4-DCA in water samples.
Environmental Water Analysis
Two water samples were collected from the East River and the Hudson River in New York City and analyzed using the proposed method. Because no target analytes was found in either sample, the water samples were fortified by spiking chloroaniline standards. Figure 6 shows the chromatogram of a blank river water and a spiked river water with 100 μg L−1 CAs. The relative recovery and RSD (%) of investigated water samples are summarized in Table 3. Acceptable analyte recoveries and RSDs were obtained using the proposed procedure.

Chromatogram of hollow fiber-based LPME of a East River water; and b East River water spiked with 100 μg L−1 analytes. 1 2-CA, 2 3-CA, 3 2,3-DCA, 4 2,4-DCA, 5 3,5-DCA, 6 3,4-DCA. Extraction conditions: 6 mL sample with 1 M NaOH, no salt addition, 1,200 rpm stirring speed and 20 min extraction time
Conclusions
The present study developed a hollow fiber-based LPME method that is applicable to the determination of six trace level chloroanilines in river water samples. Low detection limits have been obtained using GC-FID owing to the high enrichment factor offered by the procedure. The method is simple, fast and sensitive.
References
Laha S, Luthy RG (1990) Environ Sci Technol 24:363–373
Dalene M, Skarping G (1985) J Chromatogr 331:321
Moffat AC, Osselton MD, Widdop B (2004) Clarke’s analysis of drugs and poisons. Pharmaceutical Press, London
Kataoka H (1996) J Chromatogr A 733:19–34
Voyksner RD, Straub R, Keever JT, Freeman HS, Hsu W-N (1993) Environ Sci Technol 27:1665–1672
Games LM, Hites RA (1977) Anal Chem 49:1433–1440
Faraji H, Tehrani MS, Husain SW (2009) J Chromatogr A 1216:8569–8574
Zhang J, Lee HK (2006) J Chromatogr A 1117:31–37
Zhang J, Lee HK (2009) J Chromatogr A 1216:7527–7532
Wang XW, Luo LJ, Ouyang GF, Lin L, Tam NFY, Lan CY, Luan TG (2009) J Chromatogr A 1216:6267–6273
Payán MR, López MÁB, Fernández-Torres R, Bernal JLP, Mochón MC (2009) Anal Chim Acta 653:184–190
Saleh A, Yamini Y, Faraji M, Shariati S, Rezaee M (2009) J Chromatogr B 877:1758–1764
Sarafraz-Yazdi A, Mofazzeli F, Es’haghi Z (2009) Talanta 79:472–478
Rasmussen KE, Pedersen-Bjergaard S, Krogh M, Ugland HG, Gronhaug T (2000) J Chromatogr A 873:3–11
Pedersen-Bjergaard S, Rasmussen KE (2000) Electrophoresis 21:579–585
Lambropoulou DA, Albanis TA (2007) J Biochem Biophys Methods 70:195–228
Zhao LM, Lee HK (2002) Anal Chem 74:2486–2492
Shen G, Lee HK (2002) Anal Chem 74:648–654
Zhao LM, Zhu LY, Lee HK (2002) J Chromatogr A 963:239–248
Zhao RS, Yuan JP, Li HF, Wang X, Jiang T, Lin JM (2007) Anal Bioanal Chem 387:2911–2915
Es’haghi Z (2009) Anal Chim Acta 641:83–88
Pardasani D, Kanaujia PK, Gupta AK, Tak V, Shrivastava RK, Dubey DK (2007) J Chromatogr A 1141:151–157
Sarafraz-Yazdi A, Es’haghi Z (2006) Chromatographia 63:563–569
Peng J-F, Liu J-F, Jiang G-B, Tai C, Huang M-J (2005) J Chromatogr A 1072:3–6
Jeannot MA, Przyjazny A, Kokosa JMJ (2010) Chromatogr A 1217:2326–2336
Pan L, Adams M, Pawliszyn J (1995) Anal Chem 67:4396
Huang S-P, Huang SD (2007) J Chromatogr A 1176:19–25
Skoog DA, Holler FJ, Crouch SR (2007) Principles of instrumental analysis, 6th edn, Thomson Brooks/Cole, Belmont, USA
Hou L, Lee HK (2003) Anal Chem 75:2784–2789
Acknowledgments
Support for undergraduate research training provided by US Department of Education Title V grant for institutional development to John Jay College and the PRISM program at John Jay College are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Young, A., Lai, G., Hung, B. et al. Determination of Trace Chloroanilines in Environmental Water Samples Using Hollow Fiber-Based Liquid Phase Microextraction. Chromatographia 74, 83–88 (2011). https://doi.org/10.1007/s10337-011-2022-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-011-2022-6