
Abstract
Studies of Atlantic forest (AF) organisms suggest that the historical dynamics of the forest cover produced demographically stable populations in its central region and unstable populations in the southern regions. We studied the mitochondrial phylogeographic structure of an AF passerine, the Greenish Schiffornis Schiffornis virescens (Tityridae), and evaluated questions related to the history of the AF. We analyzed cytochrome b and control region sequences of the mitochondrial genome by traditional phylogenetic and population genetic methods based on summary statistics. In addition, we used coalescent simulations to evaluate specific models of evolution of the populations of S. virescens. The results did not support phylogeographic partitions of the genetic variability of S. virescens. The overall Φst was = 0.32 and gene flow between regions was moderate to high. The analyses suggested that the total population of S. virescens suffered a bottleneck followed by a demographic expansion in the late Pleistocene. The bottleneck might have contributed to the extinction of intraspecific lineages, and hence to the observed lack of a strong phylogeographic pattern and low genetic diversity. Our results suggest that some AF taxa have had all their populations similarly affected by the recent history of the biome, contrary to what has been revealed from most of the other phylogeographic studies in the region and as suggested by a model of AF refuges (the Carnaval–Moritz model). We suggest that the response of organisms to common histories may be idiosyncratic, and predictions about the history of the biome should take into account ecological characteristics and distribution of each specific taxa.
Zusammenfassung
Matrilineare Hinweise auf demografische Expansion, geringe genetische Diversität und fehlende phylogeografische Struktur bei der Olivtrauerkotinga Schiffornis virescens (Aves: Tityridae), einer endemischen Vogelart der Mata Atlantica
Untersuchungen an Organismen der Mata Atlantica deuten darauf hin, dass die historische Dynamik der Waldbedeckung dort demografisch stabile Populationen in den inneren Bereichen und instabile Populationen in den südlichen Regionen hervorgebracht hat. Wir betrachteten die mitochondriale phylogeografische Struktur bei der Olivtrauerkotinga S. virescens (Tityridae), einem Sperlingsvogel der Mata Atlantica, und untersuchten Fragen im Zusammenhang mit der Geschichte dieses atlantischen Waldes. Wir analysierten Sequenzen von Cytochrom b und von Kontrollregionen des mitochondrialen Genoms unter Anwendung der gebräuchlichen phylogenetischen und populationsgenetischen Methoden auf der Grundlage statistischer Parameter. Zusätzlich verwendeten wir Coalescent Simulations, um spezifische Evolutionsmodelle für die Populationen von S. virescens zu testen. Die Ergebnisse konnten keine phylogeografischen Untereinheiten in der genetischen Variabilität von S. virescens belegen. Der Gesamtwert für Φst betrug 0,32 und der Genfluss zwischen den Regionen war mäßig bis hoch. Die Analysen deuten an, dass die gesamte Population von S. virescens zunächst durch einen genetischen Flaschenhals ging, worauf anschließend im Oberen Pleistozän eine demografische Expansion erfolgte. Dieser Flaschenhals könnte zum Aussterben intraspezifischer Abstammungslinien und somit zum beobachteten Mangel an deutlichen phylogeografischen Mustern und zu der geringen genetischen Diversität beigetragen haben. Unsere Ergebnisse legen nahe, dass bei manchen Taxa der Mata Atlantica alle Populationen durch die jüngere Geschichte dieses Bioms ähnlich beeinflusst wurden. Dies steht im Gegensatz zu den Erkenntnissen der meisten anderen phylogeografischen Studien aus dieser Region und zu einem Modell atlantischer Waldrefugien (Carnaval–Moritz-Modell). Wir vermuten, dass verschiedene Organismen selbst auf eine gemeinsame Geschichte in ihrer ganz eigenen Weise reagieren. Prognosen über die Geschichte des Bioms sollten daher immer auch die ökologischen Eigenschaften und die Verbreitung jedes einzelnen Taxons berücksichtigen.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Atlantic forest (AF) of Brazil, northeastern Argentina and eastern Paraguay is amongst the richest and most endangered rainforests in the world (Galindo Leal and de Gusmão Câmara 2003) (Fig. 1). Congruence among palynological studies (i.e. Behling 2002; Behling and Negrelle 2001; Ledru et al. 2005), and models of paleo-distribution of forests and of some species of frogs (Carnaval and Moritz 2008; Carnaval et al. 2009), suggest that during the late Pleistocene the southern AF was strongly fragmented by the advance of grasslands and of other savanna-like vegetation. In contrast, the central and northern AF were relatively stable. More precisely, during the last glacial maximum (between 24,000 to 18,000 before present; Anderson et al. 2007), the southern grassland–forest ecotone seemed to have shifted 750 km north from its current location, which was possibly established in the late Holocene (Behling 2002). Under the former scenario of temporal and geographic stability–instability of the AF (Carnaval and Moritz (2008), hereafter the Carnaval–Moritz model, the majority of the forest-dependent taxa should have been similarly affected by the retraction and fragmentation of forests during the Pleistocene, and therefore the existence of shared phylogeographic patterns is expected.

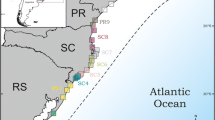
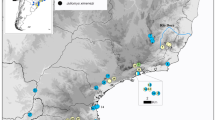
a Distribution of the Atlantic forest (AF), of Schiffornis virescens and of the sampling localities for the genetic study. See “Appendix 1” for details of sampling localities. Phylogeographic barriers observed in other AF organisms are: barrier I, the passerine Sclerurus scansor (d’Horta et al. 2011), the passerine Xiphorhynchus fuscus (Cabanne et al. 2008), the passerine Pyriglena leucoptera (Maldonado-Coelho 2012), frogs of the genus Hypsiboas (Carnaval et al. 2009), and the toad Rhinella crucifer (Thomé et al. 2010); barrier II, opossum Marmosops incanus (Mustrangi and Patton 1997), the passerines Sclerurus scansor (d′Horta 2009) and Conopophaga lineata (Pessoa et al. 2006; Pessoa 2007); barrier III, the passerines Xiphorhynchus fuscus (Cabanne et al. 2007, 2008) and Sclerurus scansor (d’Horta et al. 2011), frogs of the genus Hypsiboas (Carnaval et al. 2009), and monkeys of genus Alouatta (Martins et al. 2011); barrier IV, the passerine Dendrocolaptes platyrostris (Cabanne et al. 2011), the bee Melipona quadrifasciata (Batalha-Filho et al. 2010), the viper Bothrops jararaca (Grazziotin et al. 2006), the frogs Hypsiboas faber (Carnaval et al. 2009), Proceratophrys boiei (Amaro et al. 2012) and another frog of the genus Thoropa (Fitzpatrick et al. 2009). b Predicted phylogeographic pattern in S. virescens based on the genetic constitution of other AF species (see a). The predicted genetic structure among regions (Φct > 0.2) is related to a level of gene flow that is not sufficient to avoid allele fixation and strong divergence (Wang 2004)
The Carnaval–Moritz model has received particular support from some phylogeographic studies with forest birds (i.e. Maldonado-Coelho 2012; Cabanne et al. 2008; d’Horta et al. 2011), frogs (Carnaval et al. 2009) and bees (Batalha-Filho et al. 2010). But, on the other hand, the model has been recently challenged by a phylogeographic study of the Rhinella crucifer group of toads (Thomé et al. 2010) and by a study of planarians (Álvarez-Presas et al. 2011), which did not find support for a southern AF Holocene colonization and therefore suggest persistence of forested habitats in the south (a southern refuge). In addition, the existence of a southern refuge is further supported by a recent study with species distribution models of several endemic taxa (Porto et al. 2012).
In the present study, we used sequence data derived from mitochondrial DNA (mtDNA) to explore the population genetic structure of the Greenish Schiffornis Schiffornis virescens (Tityridae; Remsen et al. 2009) (Fig. 1a) and to address questions of the AF history. S. virescens is a good model to address issues related to the AF history because it is endemic to the biome and occurs with low to medium density in well-preserved forests (Stotz et al. 1996). The species does not present geographic variation in plumage, song or morphometry (Ridgely and Tudor 1996). Since it inhabits the understory of preserved forests and is sensitive to forest fragmentation (Anjos et al. 2011; Stotz et al. 1996), chances to have been affected by Pleistocene fragmentation of habitat, and therefore to present a strong population genetic structure, are high (Burney and Brumfield 2009). Assuming that most AF organisms were affected similarly by the Pleistocene forest dynamics, we expected to find in S. virescens results similar to those obtained by other studies of AF organisms, which apparently revealed a shared phylogeographic pattern (Maldonado-Coelho 2012; Batalha-Filho et al. 2010; Grazziotin et al. 2006; Cabanne et al. 2007, 2008; Mustrangi and Patton 1997; Carnaval et al. 2009; d’Horta et al. 2011; Pessoa 2007). This shared pattern for each species consists of the existence of two main mtDNA clades south of the Doce river that come into contact in south-central São Paulo where no barrier to gene flow is evident (Fig. 1a, b). In addition, some species present a third lineage north of the Doce river (Cabanne et al. 2008; d’Horta et al. 2011; Maldonado-Coelho 2012). Another characteristic of the shared phylogeographic pattern is that the corresponding southern clades present evidence of population expansion, differently to clades north of the Doce river. These shared phylogeographic features could be the product of alterations in the forest cover that occurred during the late Pleistocene, as would be expected from the Carnaval–Moritz model.
Phylogeographic studies of AF organisms are still scant and a robust synthetic hypothesis about the evolution of AF organisms is not yet possible (but see Carnaval et al. 2009; Martins et al. 2011). Up to now, the selection of AF species for phylogeographic studies was not only driven by biogeographic questions but also by systematic issues in polymorphic taxa (i.e. Cabanne et al. 2008). This aspect may have biased the possibility of finding complex phylogeographic patterns because variation in external characters (i.e. plumage and voice) is frequently correlated with genetic structure. S. virescens has no described geographic variation in external characters, therefore an alternative working hypothesis might be to find no phylogeographic structure.
Nyari (2007) showed that S. turdina, which is the sister species of S. virescens, presents a substantial intra-specific genetic structure. Because both species are very similar in song and plumage, the evolutionary relationship between them is not yet clear. The study of Nyari (2007) included samples of S. virescens, but without sampling all the variation. Thus, before addressing our biogeographic questions, we evaluated whether the species is monophyletic.
The objective of this study was to describe the mtDNA genetic variation of S. virescens to address the following questions: (1) Is the species monophyletic? (2) Does it present the phylogeographic structure described for other AF organisms? (3) How did the species’ phylogeographic pattern originate? and (4) What are the implications of results for understanding the evolution of AF organisms?
Methods
Genetic datasets
In the phylogenetic analysis, we included 28 S. virescens, 14 S. turdina representing the variation of that species (Nyari 2007), as well as 1 S. major and 2 Laniocera hypopyrra as the outgroup. We used an alignment consisting of 840 bp of the cytochrome b (cytb) and 476 bp of the first domain of the mtDNA control region (CR). Sequences of cytb from S. turdina, two S. virescens and the two outgroups were obtained from the study of Nyari (2007) (see GenBank accession numbers in Fig. 2). We were unable to collect CR sequences for some S. turdina and for the outgroups, so we coded these as missing data. Amplification and sequencing of cytb was done following Cabanne et al. (2008). See “Appendix 1” for details of the samples used in this study.

a Demographic models of Schiffornis virescens evaluated by coalescent simulations in BAYESSC. We simulated populations (shown in black) that suffered different historical events (He): stability (A), bottlenecks (B–D), expansion (E), and the evolution in an island system (F, G). b Details of historical events and population parameters used in simulations. The prior distribution of time of events (t), migration (m) and of the demographic growth rate (r) are specified. Continuous priors are denoted between parentheses. Normally distributed priors are denoted by their mean and standard distribution (SD). Intensities of bottlenecks are indicated by the proportion of the current effective size (Ne) that persisted during the bottleneck. Time is expressed in kilo-years (ky). LGM last glacial maximum
For the population genetic study, we used CR sequences (476 pb) from 57 S. virescens collected in 19 localities (Fig. 1a). Amplification and sequencing of the CR was performed by specifically designed primers: forward LSvCB GATCTATCCCCAATAAACTTGGAGG and reverse HSvCR GATTACGAACTCGAAGTTGGACGG. PCR annealing temperature for CR was 60 °C for S. virescens and 53 °C for S. turdina. In order to avoid working with possible copies of the CR translocated to the nucleus, we only used sequences obtained from clean amplification bands, presenting the expected length, single peaks, and a base composition compatible with mtDNA (Baker and Marshall 1997).
Since both cytb and CR are physically linked in a single molecule (mitochondrial genome), results obtained with both markers are totally compatible because they reflect a single genealogy and history (Avise 2000). Genbank accession numbers for cytb and for CR are JX477687–JX477770. For evaluating whether the used sequences were representative of the genetic constitution of S. virescens, we estimated P = [(k − 1)/(k + 1)], which represents the probability that a sample of size k and the whole population share the most recent common ancestor (Hein et al. 2005). Hence, P can be interpreted as the probability of the sample being representative of the genetic diversity of the population.
Phylogenetic analyses and molecular clock
Molecular evolution parameters were selected in MODELTEST 3.7 (Posada and Crandall 1998) or directly estimated during phylogenetic reconstruction. To obtain phylogenetic trees, we used bayesian and maximum likelihood approaches as implemented in MrBAYES 3.1.2 (Ronquist and Huelsenbeck 2003) and PHYML 2.4.4 (Guindon and Gascuel 2003), respectively. MrBAYES analysis used a MCMC of 5 × 106 generations with sampling every 1,000 generations. We discarded the first 1,750 genealogies (burn-in), and parameters of molecular evolution between genes (cytb and CR) were unlinked. CR sequences of some taxa were coded as missing data. As missing data might potentially mislead phylogenetic inference (Lemmon et al. 2009), we analyzed the dataset with and without CR sites. As topologies did not change, the final phylogenetic analyses used the dataset with missing data (cytb+CR).
We used BEAST 1.6.1 (Drummond and Rambaut 2007) to estimate node ages by using a relaxed uncorrelated lognormal molecular clock analysis based on the cytb partition, a molecular clock calibration of 2.1 % divergence per million years (Weir and Schluter 2008), and a tree prior of constant size. From this analysis in BEAST, we estimated a substitution rate for the CR in relation to the cytb rate. Node ages obtained with the cytb-CR dataset (with missing data) and with only the cytb (no missing data) were similar, and therefore we based our dating estimation in the cytb-CR dataset.
To study the constancy of the molecular clock we used two procedures. First, we evaluated the parameter ucld.stdev (standard deviation of the molecular rate of cytb), estimated in BEAST. If the parameter estimate is close to zero then the data is clock-like. If the ucld.stdev has an estimated value much greater than one, then the data exhibits substantial rate heterogeneity among lineages (Drummond and Rambaut 2007). Secondly, we used a likelihood-ratio test, as implemented in MODELTEST, by comparing the log-likelihood values of maximum likelihood trees with and without enforcing a molecular clock.
Lastly, visualization of relationships among CR haplotypes was done by a median-joining network constructed in NETWORK 4.1.0.8 (http://www.fluxus-engineering.com).
Population genetic analysis
All the population genetic analyses used only the CR dataset because this marker was more variable than the cytb. To test the existence of phylogeographic barriers (gene flow barriers between populations), we used analyses of molecular variance (AMOVA) in ARLEQUIN 3.1 (Excoffier et al. 2006) based on uncorrected distances. Specifically, we evaluated the Φct statistic, which represents the proportion of the total molecular diversity variance that is explained by genetic differences among geographic regions. These regions were delimited by the hypothetical phylogeographic barriers (Fig. 1a). Φ ct = Va/Vt, where Va is the molecular variance among groups and Vt is the total molecular variance (Excoffier et al. 1992). We also evaluated Φ st = (Va + Vb)/Vt, where Vb is the molecular variance among populations within groups (Excoffier et al. 1992). Significance and confidence intervals of fixation indices were calculated based on 20,000 permutations.
We estimated the parameter Θ by a maximum likelihood coalescent method as implemented in LAMARC 2.1.2b (Kuhner 2006). We also used LAMARC to evaluate the demographic signature by estimating the exponential population growth rate g of the model Θ t = Θnowe−gt, where Θnow is the current Θ and Θ t is the value of the parameter t time ago. Positive g values indicate population growth and negative values indicate population decline. Runs used the F84 model of sequence evolution with empirical base frequencies and transition/transversion ratios, and default characteristics of the Markov Chain Montecarlo.
We also evaluated the historical demography using the neutrality tests of Tajima (1989) and of Fu (1997) in DNAsp 5.1 (Librado and Rozas 2009). If the marker is selectively neutral (not positively selected), significant negative values of the test statistics suggest demographic expansion. In addition, to evaluate the hypothesis of positive selection, we used the compound test HEW and DHEW (Zeng et al. 2007), performed with S. turdina as outroup in the NH software (kindly provided by K. Zeng). The tests HEW and DHEW evaluate the null hypothesis of neutrality and are significant only when positive selection has acted in the target sequence or linked regions (hitchhiking) (Zeng et al. 2007). The skyride analysis (Minin et al. 2008) for studying the change of population effective size through time was performed in BEAST 1.6.1 with 10 million generations, sampling every 1,000 generations and a burn-in of 1,000 genealogies. We used an uncorrelated log-normal clock model, time aware smoothing and a generation time of 1 year (Klicka and Zink 1999).
Estimation of gene flow assuming an island model in equilibrium was obtained using the formula M = (1 − Φst)/2Φst (Hedrick 2000). Also, we used the isolation–migration (IM) model (Nielsen and Wakeley 2001; Hey and Nielsen 2004), implemented in IM (v. April 21, 2008), to estimate non-equilibrium gene flow rates and divergence times between regions. We applied the HKY model of evolution, similar population parameter theta values (Θ1 = Θ2 = Θ a ) and m 1 = m 2 . A total of 500,000 iterations were used for burn-in and analyses were stopped when the smallest effective sample size (ESS) was higher than 50 and parameter trend lines stabilized, usually after 20 million iterations. We assumed a generation time of 1 year.
Coalescent simulations
In order to evaluate different demographic histories, we simulated demographic scenarios in the program BAYESSC, a modification of software SERIAL SIMCOAL (Anderson et al. 2005; Chan et al. 2006), and evaluated the goodness of fit of the observed data (CR sequences) to the simulations; see Richards et al. (2007) and Knowles (2009) for reviews on this approach. Because genetic signals of expansion may be found in a demographically stable island system (Nielsen and Beaumont 2009; Peter et al. 2010), the main objective of this section was to investigate if the phylogeographic pattern could have been generated in an island system with demographic stability or in a non-structured system that expanded demographically. The demographic models assessed, detailed in Fig. 2, differed in the timing, intensity, and numbers of bottlenecks, and also in the number of populations. The populations in model G were originated before the Pleistocene, as suggested by the divergence of S. virescens with its sister species (see “Results”). In order to obtain a metric of population genetic structure (Φst) for the models of single populations and to simulate the island models, we organized the sampling localities by geographic proximity into five populations (pop). This procedure is a conventional practice in this kind of analysis (i.e. Fabre et al. 2009; Belle et al. 2006). In preliminary analyses, we tested several other population configurations and results did not change. The final organization of sampling localities was: pop 1: localities 1–5; pop 2: localities 6–8, 11; pop 3: localities 12–14; pop 4: localities 9, 10, 15–17; pop 5: localities 18 and 19. See localities in “Appendix 1”.
For each model, we performed 1,000 simulations and used ARLEQUIN to estimate five summary statistics for the complete dataset: number of segregating sites, nucleotide diversity, Tajima’s D (Tajima, 1989), Fu’s Fs (Fu 1997), and Φst amongst populations (Excoffier et al. 1992). To evaluate the plausibility of each model, we first estimated the proportion pi of simulated values equal and higher than the observed summary statistics, and then we reported a global p value for each model by combining individual pi values by a nonparametric procedure described in Voight et al. (2005) and Fabre et al. (2009). See electronic supplementary material for further methodological details.
Results
Genetic data, neutrality and mtDNA diversity
The cytb and CR concatenated alignment and the CR alignment presented probabilities to represent the complete gene genealogy equal to 93 and 96.5 %, respectively, which indicated that the samples are suitable to the study. The phylogenetic analysis showed that S. virescens is a well-supported monophyletic lineage (Fig. 3a) that diverged from its sister species, S. turdina, approximately 6.2 Mya (million years ago) (HPD95 % 4.3–8.14 Mya). Contrary to what we hypothesized, no intraspecific matrilines genetic structure was present in S. virescens. The CR dataset presented 17 haplotypes and the network confirmed the lack of phylogeographic structure (Fig. 3b). The parameters cytb.ucld.stdev = 0.293 (HPD95 % 0.0000015–0.76) and CR.ucld.stdev = 0.3757 (HPD95 % 0.0000119–1.05) estimated in BEAST indicated a slight deviation from the constant molecular clock in cytb and CR sequences, respectively. This result was further confirmed by the likelihood ratio test for the dataset S. virescens/S. turdina and for the datasets of each species (p < 0.00001). The divergence rate of CR estimated in BEAST was 2.34 % divergence per million years, slightly higher than the cytb rate (2.1 % divergence per million years). We used this rate in the following analyses with CR.

a Bayesian phylogenetic tree based on 1,533 bp of the cytb and control region of the mtDNA of Shiffornis virescens and closely related species. Each gene partition used the model of molecular evolution GTR+I+G with parametes specifically estimated. Posterior probability obtained in MrBAYES (Bay. prob) and maximum likelihood bootstrap (ML Boot, 500 replicates) are presented at nodes. Divergence dates were obtained in BEAST based in the cytb molecular clock (Weir and Schluter 2008). b Median joining network based on 476 bp of the mtDNA control region of S. virescens. c Skyride plot showing the variation in population effective (Ne) size through time
We used cytb to compare the genetic diversity of S. virescens with other AF birds. We reviewed the available studies on AF birds and found that the nucleotide diversity of a sample of 12 species (including S. virescens) ranged from 0.01574 to 0.00187 (average = 0.0086, see “Appendix 2” for details). S. virescens presented the smallest diversity (cytb nucleotide diversity = 0.00187) among the 12 species considered.
The AMOVA results did not support any of the hypothesized genetic barriers (Table 1). All Φct values were small and not significant, which indicates that most of the variation in the mtDNA of S. virescens was organized among and within populations, and not among regions separated by the tested barriers. In addition, the global Φst value (Φst = 0.3184) indicated a moderate population genetic structure among localities, which corresponded to moderate equilibrium gene flow of ~1.1 females per generation among populations. The analysis in IM indicated moderate to high gene flow between regions separated by the putative phylogeographic barriers (Fig. 1a): M CAF–SAF: 0.8 females (HPD95 % 0–6.5), M CAF–SEAF: 5.5 females (HPD95 % 1.6–42.7); and M SAF–SEAF: 10.4 females (HPD95 % 1.7–?). All divergence times correspond to the late Pleistocene: T CAF–SAF: 0.16 MYA (HPD95 % 0.04–0.7), T CAF–SEAF: 0.1 MYA (HPD95 % 0.0014–0.5); and T SAF–SEAF: 0.06 MYA (>0.012). However, as all posterior distributions of divergence estimations are very wide (results not shown), populations cannot be considered as independent, which confirms the absence of population structure in S. virescens.
The summary statistics tests collectively suggested a demographic expansion in S. virescens (Table 2). The neutrality–demographic tests suggested a demographic expansion because Fu’s tests were significant. Results of the test of Tajima were borderline but compatible with the Fu’s tests because the D statistics were negative. Results of the positive selection tests did not support the action of positive selection; however, some p values are borderline significant. The demographic analysis in LAMARC further corroborated that S. virescens expanded recently (positive g = 932, HPD95 % CI: 367–1966). The Skyride plot analysis showed the existence of a moderate expansion during the late Pleistocene, near 21 times the initial Ne (Fig. 3c). The region of origin of the demographic expansion should present the highest level of genetic diversity. However, because the three regions into which we divided the study area (Fig. 1a; CAF, SEAF and SAF) presented similar levels of genetic diversity (results not shown), the origin of the expansion was not detected.
Simulations results
Results of the evaluation of demographic models by coalescent simulations are presented in Table 3. The model A of panmixia and demographic stability, the model E of panmixia and expansion, and the models of islands and demographic stability F and G obtained low global probabilities, indicating they all have low plausibility. All the models with high global probability (p value >0.1: models B, C, and D) suggest that S. virescens suffered bottlenecks during the glacial maxima followed by posterior recovering of the population effective size. In particular, the model with highest p value (model C1, p = 0.84) suggests that S. virescens is conformed by a single population that suffered an intense bottleneck before the last glacial maxima.
The results of these simulations are compatible with the analysis performed in LAMARC, as well as with the demographic summary statistics and the skyride plot, collectively suggesting that the species suffered an expansion during the late Pleistocene. A genetic pattern compatible with demographic expansion could also result from an island system with demographic stability, but our simulations did not support this situation (models F and G).
Discussion
Schiffornis virescens was found to be monophyletic, and presented low mtDNA genetic diversity and a weak population genetic structure. In addition, the genetic partitions across genetic barriers important for other organisms were not found to be influential for S. virescens. These results are in contrast to what was found in its sister species, S. turdina, and in other AF taxa dependent on preserved forests. S. turdina has high genetic diversity and substantial phylogeographic structure (Nyari 2007). Within the lineage S. virescens/S. turdina, the most ancient split separated birds from the AF (S. virescens) from birds associated primarily to the Amazon and Central America forests (S. turdina). Interestingly, and as observed in some other taxa (i.e. Cabanne et al. 2008; Ribas et al. 2005), the AF lineage showed shorter branches and lower genetic distances among terminals (Fig. 3a), a pattern that suggests different evolutionary dynamics in both Schiffornis species (Ricklefs 2007). Also, contrary to what was expected according to other Neotropical rainforest organisms from the AF or from outside the AF (i.e., Cabanne et al. 2011; d’Horta et al. 2011; Carnaval et al. 2009; Bates 2002; Aleixo 2004; Cheviron et al. 2005; Nyari 2007), S. virescens did not present a strong phylogeographic structure.
The differences in matrilineal population structures among birds could not be explained by ecological divergence between S. virescens and the other AF birds. Ecology predicts levels of intraspecific genetic divergence in birds from Amazonia and likely from other regions (Burney and Brumfield 2009). For example, canopy birds tend to be less genetically differentiated than understory birds. In addition, very specialized bird taxa are more diversified than generalist birds (Salisbury et al. 2012). Even though comparative ecological studies of AF birds are scarce (i.e., Anjos 2006; Ribon et al. 2003), there are not remarkable ecological differences between S. virescens and the AF birds that present strong phylogeographic structure (Cabanne et al. 2008, 2011; d’Horta et al. 2011; Maldonado-Coelho 2012; Pessoa et al. 2006; Lacerda et al. 2007). S. virescens, as well as most of the other studied birds, inhabits the understory and is a specialist of well-preserved forests. A pattern of low genetic structure, such as the one found in S. virescens, is expected for species with high gene flow and low sensitivity to forest fragmentation. An example of such a species is Basileuterus leucoblepharus (Anjos 2006), which is a bird co-distributed with S. virescens whose mtDNA and nuclear DNA are not geographically structured and that presented demographic stability during the late Pleistocene (Batalha-Filho et al. 2012).
The mtDNA genealogical pattern of low diversity and no genetic structure of S. virescens could have been generated by a selective sweep (positive selection) (Nielsen 2005). However, we utilized specific tests for positive selection, HEW and DHEW, and the results did not support the hypothesis of selection (Table 2). Therefore, the genetic constitution of S. virescens could have been modeled by factors other than positive selection, such as demographic processes.
S. virescens is a relatively young species, as shown by its time to the most recent common ancestor (TMRCA) (0.43 Mya; Fig. 3a), and thus not enough time may have passed for the evolution of intraspecific structure. However, S. virescens is not younger than other AF species that do present a strong phylogeographic structure, such as Sclerurus scansor and Dendrocolaptes platyrostris (d’Horta et al. 2011; Cabanne et al. 2011), therefore species age alone cannot account for the lack of phylogeographic structure.
The difference between the splitting time between S. virescens and S. turdina (6.2 Mya) and the TMRCA of S. virescens (0.43 Mya) seems to be high when compared to other pairs of sister taxa of the region. For example, in other AF species such as S. scansor (d’Horta et al. 2011) and Xiphorhynchus fuscus (Cabanne et al. 2008), the divergence with their sister clades are ~1.1 and ~3 Mya ago, respectively, while the maximum intraspecific divergence are about 0.6 and 0.8 Mya, respectively. Also, in D. platyrostris, another passerine from the AF and the gallery forests of central South America savannas, the divergence with its sister taxa is about 1.6 Mya while the intraspecific divergence is 0.22 Mya (Cabanne et al. 2011). We expected a higher intraspecific differentiation in S. virescens, and thus an older TMRCA, because of the old splitting time with S. turdina and because of the high intraspecific diversity documented within the latter species. The matrilineal constitution of S. virescens suggests an evolutionary history that resulted in low intraspecific diversification. Perhaps, lower diversification rates or higher extinctions rates in S. virescens could be responsible for this pattern (Ricklefs 2007).
The phylogeographic pattern of S. virescens could have been originated through the combined effects of gene flow and historical demography. Our results indicated that gene flow rates among populations are moderate to high, enough to avoid allele fixation and strong divergence (Wang 2004). Also, different analyses collectively revealed a recent population expansion. Finally, S. virescens presented lower levels of mtDNA genetic diversity than other AF birds (“Appendix 2”), suggesting that the demographic expansion may have occurred after a population bottleneck that reduced genetic diversity. This bottleneck might have contributed to the extinction of intraspecific mtDNA lineages, leading to the observed lack of a strong phylogeographic pattern and low genetic diversity in S. virescens.
Different effective population sizes, demography, or the lack of recognition of different species in S. turdina might be responsible for the differences in mtDNA genetic diversity and structure between S. virescens and S. turdina. S. turdina inhabits all the Amazonian basin forests, part of Central America forests, and part of northeastern AF. Thus, its geographic range is nearly five times larger than that of S. virescens, which may imply a larger population and therefore higher genetic diversity for S. turdina. In addition, S. turdina has several highly divergent phylogroups that, with further study, might be considered as good species (Nyari 2007).
The phylogeographic pattern of a species can be shaped by the history of the biome in which it is found (Hewitt 2000, 2004). Our results, specifically the lack of a strong phylogeographic structure, the low genetic diversity, a most recent common ancestor in the mid-Pleistocene, and the evidence of demographic expansion for the whole species, suggest that the Ne of S. virescens was reduced greatly during the late Pleistocene, likely to a population restricted to a single continuous forest (refuge) of uncertain location. Interestingly, the magnitude of the demographic expansion of S. virescens (~21 × initial Ne; Fig. 3c) is similar to that described for some taxa from the northern hemisphere that were highly affected by the cycles of forest shrinkage—expansion that occurred during global glaciations (e.g., Catharus ustulatus; Ruegg et al. 2006).
Most populations of S. virescens inhabit a region whose rainforest coverage fluctuated during the Pleistocene, which could be the reason why they presented a shared demographic history. Specifically, the majority of the range of the bird is located in the southern portion of the AF, which was the most affected by natural fragmentation and expansion of savanna-like vegetation during the global glaciations (Carnaval et al. 2009; Behling 2002; Behling et al. 2002). This is different from those taxa that persisted in the same period in more than one population, and whose geographic ranges also span historically stable forests in central and northern AF (e.g., d’Horta et al. 2011; Cabanne et al. 2008, 2011; Thomé et al. 2010).
Our results were not fully compatible with the predictions derived from the Carnaval–Moritz model (Fig. 1b), but they were still in accordance with some aspects of the hypotheses of the AF history. Most published studies on AF organisms suggest that the Pleistocene dynamics produced stable populations in the central region and unstable populations in the southern portion. We did not recover this pattern in S. virescens, but rather documented that its association to the southern AF and relatively short age was crucial for this resulting pattern. The Carnaval–Moritz model is not powerful enough to predict the distribution of Pleistocene refugia of organisms restricted to the southeast–south portion of the AF (Porto et al. 2012), and our results suggested that its power to predict phylogeographic patterns of those organisms is also low. Independent of the origin of the phylogeographic structure within S. virescens, our results indicated that some AF taxa have had all their extant populations similarly affected by the recent history of the biome, contrary to what has been revealed from most of the other phylogeographic studies of species from the region. Therefore, the response of organisms to common histories may be idiosyncratic, and predictions about the history of the biome should take into account ecological characteristics and distribution of each specific taxa.
References
Aleixo A (2002) Molecular systematics and the role of the “varzea” - “terra-firme” ecotone in the diversification of Xiphorhynchus woodcreepers (Aves : Dendrocolaptidae). Auk 119(3):621–640
Aleixo A (2004) Historical diversification of a Terra-firme forest bird superspecies: a phylogeographic perspective on the role of different hypotheses of Amazonian diversification. Evolution 58(6):1303–1317
Álvarez-Presas M, Carbayo F, Rozas J, Riutort M (2011) Land planarians (Platyhelminthes) as a model organism for fine-scale phylogeographic studies: understanding patterns of biodiversity in the Brazilian Atlantic Forest hotspot. J Evol Biol 24(4):887–896
Amaro RC, Carnaval AC, Yonenaga-Yassuda Y, Trefaut Rodrigues M (2012) Demographic processes in the montane Atlantic rainforest: molecular and Cytogenetic evidence from the endemic frog Proceratophrys boiei. Mol Phylogenet Evol 63:880–888
Anderson CNK, Ramakrishnan U, Chan YL, Hadly EA (2005) Serial SimCoal: a population genetics model for data from multiple populations and points in time. Bioinformatics 21(8):1733–1734
Anderson D, Goudie A, Parker A, Goudie A (2007) Global environments through the Quaternary: exploring environmental change. Oxford University Press, Oxford
Anjos L (2006) Bird Species Sensitivity in a Fragmented Landscape of the Atlantic Forest in Southern Brazil1. Biotropica 38(2):229–234
Anjos L, Collins CD, Holt RD, Volpato GH, Mendonça LB, Lopes EV, Boçon R, Bisheimer MV, Serafini PP, Carvalho J (2011) Bird species abundance-occupancy patterns and sensitivity to forest fragmentation: implications for conservation in the Brazilian Atlantic forest. Biol Conserv 144:2213–2222
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge
Baker AJ, Marshall HD (1997) Mitochondrial control region sequences as tools for understanding evolution. In: Mindell DP (ed) Avian molecular evolution and systematics. Academic, San Diego, pp 51–82
Batalha-Filho H, Waldschmidt AM, Campos LAO, Tavares MG, Fernandes-Salomão TM (2010) Phylogeography and historical demography of the Neotropical stingless bee Melipona quadrifasciata (Hymenoptera, Apidae): incongruence between morphology and mitochondrial DNA. Apidologie 41(5):534–547
Batalha-Filho H, Cabanne GS, Miyaki CY (2012) Phylogeography of an Atlantic forest passerine reveals demographic stability through the last glacial maximum. Mol Phylogenet Evol. http://dx.doi.org/10.1016/j.ympev.2012.08.010
Bates JM (2002) The genetic effects of forest fragmentation on five species of Amazonian birds. J Avian Biol 33(3):276–294
Behling H (2002) South and southeast Brazilian grasslands during Late Quaternary times: a synthesis. Palaeogeogr Palaeoclimatol Palaeoecol 177(1–2):19–27
Behling H, Negrelle RRB (2001) Tropical rain forest and climate dynamics of the Atlantic lowland, Southern Brazil, during the late Quaternary. Quat Res 56(3):383–389
Behling H, Arz HW, Patzold J, Wefer G (2002) Late Quaternary vegetational and climate dynamics in southeastern Brazil, inferences from marine cores GeoB 3229–2 and GeoB 3202–1. Palaeogeogr Palaeoclimatol Palaeoecol 179(3–4):227–243
Belle EM, Ramakrishnan U, Mountain JL, Barbujani G (2006) Serial coalescent simulations suggest a weak genealogical relationship between Etruscans and modern Tuscans. Proc Natl Acad Sci USA 103(21):8012–8017
Burney CW, Brumfield RT (2009) Ecology predicts levels of genetic differentiation in Neotropical birds. Am Nat 174(3):358–368
Cabanne GS, Santos FR, Miyaki CY (2007) Phylogeography of Xiphorhynchus fuscus (Passeriformes, Dendrocolaptidae): vicariance and recent demographic expansion in southern Atlantic forest. Biol J Linn Soc 91(1):73–84
Cabanne GS, d’Horta FM, Sari EHR, Santos FR, Miyaki CY (2008) Nuclear and mitochondrial phylogeography of the Atlantic forest endemic Xiphorhynchus fuscus (Aves: Dendrocolaptidae): biogeography and systematics implications. Mol Phylogenet Evol 49:760–773
Cabanne GS, D’Horta FM, Meyer D, Silva JMC, Miyaki CY (2011) Evolution of Dendrocolaptes platyrostris (Aves: Furnariidae) between the South American open vegetation corridor and the Atlantic forest. Biol J Linn Soc 103(4):801–820
Carnaval AC, Moritz C (2008) Historical climate modeling predicts patterns of current biodiversity in the Brazilian Atlantic forest. J Biogeogr 35:1187–1201
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C (2009) Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science 323(5915):785–789
Chan YL, Anderson CNK, Hadly EA (2006) Bayesian estimation of the timing and severity of a population bottleneck from ancient DNA. PLoS Genet 2(4):451–460
Cheviron ZA, Hackett SJ, Capparella AP (2005) Complex evolutionary history of a Neotropical lowland forest bird (Lepidothrix coronata) and its implications for historical hypotheses of the origin of Neotropical avian diversity. Mol Phylogenet Evol 36(2):338–357
d′Horta FM (2009) Filogenia molecular e filogeografia de passeriformes (Aves): historia biogeográfica da região Neotropical com ênfase na Floresta Atlântica. PhD Thesis. Universidade de São Paulo, São Paulo
d’Horta F, Cabanne GS, Meyer D, Miyaki CY (2011) The genetic effects of Late Quaternary climatic changes over a tropical latitudinal gradient: diversification of an Atlantic Forest passerine. Mol Ecol 20:1923–1935
Drummond A, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes—application to human mitochondrial-DNA restriction data. Genetics 131(2):479–491
Excoffier L, Laval G, Schneider S (2006) Arlequin ver. 3.1. an integrated software package for population genetics data analysis. Computational and molecular population genetics Lab (CMPG), Inst. of Zoology, Univ. of Berne
Fabre V, Condemi S, Degioanni A (2009) Genetic evidence of geographical groups among Neanderthals. PLoS ONE 4(4):e5151
Fitzpatrick SW, Brasileiro CA, Haddad CFB, Zamudio KR (2009) Geographical variation in genetic structure of an Atlantic Coastal Forest frog reveals regional differences in habitat stability. Mol Ecol 18(13):2877–2896
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147(2):915–925
Galindo Leal C, Câmara I de G (2003) The Atlantic forest of South America: biodiversity status, threats, and outlook. State of the Hotspots. Island Press, Washington
Grazziotin FG, Monzel M, Echeverrigaray S, Bonatto SL (2006) Phylogeography of the Bothrops jararaca complex (Serpentes: Viperidae): past fragmentation and island colonization in the Brazilian Atlantic Forest. Mol Ecol 15(13):3969–3982
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52(5):696–704
Hedrick PW (2000) Genetics of populations, 2nd edn. Jones and Bartlett, Boston
Hein J, Schierup MH, Wiuf C (2005) Gene genealogies, variation and evolution : a primer in coalescent theory. Oxford University Press, Oxford
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405(6789):907–913
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B 359(1442):183–195
Hey J, Nielsen R (2004) Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167(2):747–760
Irestedt M, Fjeldsa J, Ericson PGP (2004) Phylogenetic relationships of woodcreepers (Aves: Dendrocolaptinae) incongruence between molecular and morphological data. J Avian Biol 35(3):280–288
Klicka J, Zink RM (1999) Pleistocene effects on North American songbird evolution. Proc R Soc Lond B 266:695–700
Knowles LL (2009) Statistical phylogeography. Annu Rev Ecol Evol Syst 40:593–612
Kuhner MK (2006) LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 22(6):768–770
Lacerda DR, Marini MA, Santos FR (2007) Mitochondrial DNA corroborates the species distinctiveness of the Planalto (Thamnophilus pelzelni Hellmayr, 1924) and the Sooretama (T. ambiguus Swainson, 1825) Slaty-antshrikes (Passeriformes: Thamnophilidae). Braz J Biol 67:873–882
Ledru MP, Rousseau DD, Cruz FW, Riccomini C, Karmann I, Martin L (2005) Paleoclimate changes during the last 100,000 yr from a record in the Brazilian Atlantic rainforest region and interhemispheric comparison. Quat Res 64(3):444–450
Lemmon AR, Brown JM, Stanger-Hall K, Lemmon EM (2009) The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Syst Biol 58(1):130–145
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452
Maldonado-Coelho M (2012) Climatic oscillations shape the phylogeographical structure of Atlantic Forest fire-eye antbirds (Aves: Thamnophilidae). Biol J Linn Soc 105:900–924
Martins FM, Gifalli-Iughetti C, Koiffman C, Harris E (2011) Coalescent analysis of mtDNA indicates Pleistocene divergence among three species of howler monkey (Alouatta spp.) and population subdivision within the Atlantic Coastal Forest species, A. guariba. Primates 52(1):77–87
Mata H, Fontana CS, Maurício GN, Bornschein MR, de Vasconcelos MF, Bonatto SL (2009) Molecular phylogeny and biogeography of the eastern Tapaculos (Aves: Rhinocryptidae: Scytalopus, Eleoscytalopus): cryptic diversification in Brazilian Atlantic Forest. Mol Phylogenet Evol 53(2):450–462
Minin VN, Bloomquist EW, Suchard MA (2008) Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol Biol Evol 25(7):1459–1471
Mustrangi MA, Patton JL (1997) Phylogeography and systematics of the Slender Mouse Opossum Marmosops (Marsupialia: Didelphidae). University of California Publications in Zoology 130
Nielsen R (2005) Molecular signatures of natural selection. Annu Rev Genet 39(1):197–218
Nielsen R, Beaumont MA (2009) Statistical inferences in phylogeography. Mol Ecol 18(6):1034–1047
Nielsen R, Wakeley J (2001) Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics 158(2):885–896
Nyari AS (2007) Phylogeographic patterns, molecular and vocal differentiation, and species limits in Schiffornis turdina (Aves). Mol Phylogenet Evol 44(1):154–164
Pessoa RO (2007) Sistemática e Biogeografia Histórica da Família Conopophagidae (Aves: Passeriformes): Especiação nas Florestas da América do Sul. Universidade de São Paulo, São Paulo
Pessoa RO, Cabanne GS, Sari EH, Santos FR, Miyaki CY (2006) Comparative phylogeography of the Rufous Gnateater (Conopophagidae) and Lesser Woodcreeper (Dendrocolaptidae): congruent history of two passerines from the south American Atlantic forest. J Ornithol 147(5):227–228
Peter BM, Wegmann D, Excoffier L (2010) Distinguishing between population bottleneck and population subdivision by a Bayesian model choice procedure. Mol Ecol 19(21):4648–4660
Porto TJ, Carnaval AC, da Rocha PLB (2012) Evaluating forest refugial models using species distribution models, model filling and inclusion: a case study with 14 Brazilian species. Divers Distrib. doi:10.1111/j.1472-4642.2012.00944.x
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14(9):817–818
Remsen JV, Cadena CD, Jaramillo A, Nores M, Pacheco JF, Robbins MB, Schulenberg TS, Stiles FG, Stotz DF, Zimmer KJ (2009) A classification of the bird species of South America. American Ornithologists’ Union. http://www.museum.lsu.edu/~Remsen/SACCBaseline.html. Accessed 29 Jan 2009
Ribas CC, Gaban-Lima R, Miyaki CY, Cracraft J (2005) Historical biogeography and diversification within the Neotropical parrot genus Pionopsitta (Aves: Psittacidae). J Biogeogr 32(8):1409–1427
Ribon R, Simon JE, De Mattos GT (2003) Bird extinctions in Atlantic forest fragments of the Viçosa region, southeastern Brazil. Conserv Biol 17(6):1827–1839
Richards CL, Carstens BC, Knowles LL (2007) Distribution modelling and statistical phylogeography: an integrative framework for generating and testing alternative biogeographical hypotheses. J Biogeogr 34(11):1833–1845
Ricklefs RE (2007) Estimating diversification rates from phylogenetic information. Trends Ecol Evol 22(11):601–610
Ridgely RS, Tudor G (1996) The birds of South America: the Suboscine Passerines, 1st edn. University of Texas Press, Austin
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19(12):1572–1574
Ruegg KC, Hijmans RJ, Moritz C (2006) Climate change and the origin of migratory pathways in the Swainson’s thrush, Catharus ustulatus. J Biogeogr 33(7):1172–1182
Salisbury CL, Seddon N, Cooney CR, Tobias JA (2012) The latitudinal gradient in dispersal constraints: ecological specialization drives diversification in tropical birds. Ecol Lett 15:847–855
Stotz DF, Fitzpatrick JW, Parker TA III, Moskovits DK (1996) Neotropical birds: ecology and conservation. University of Chicago Press, Chicago
Tajima F (1989) Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595
Thomé MTC, Zamudio KR, Giovanelli JGR, Haddad CFB, Baldiserra FA, Alexandrino J (2010) Phylogeography of endemic toads and post-Pliocene persistence of the Brazilian Atlantic Forest. Mol Phylogenet Evol 55:1018–1031
Voight BF, Adams AM, Frisse LA, Qian Y, Hudson RR, Di Rienzo A (2005) Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc Natl Acad Sci USA 102(51):18508–18513
Wang JL (2004) Application of the one-migrant-per-generation rule to conservation and management. Conserv Biol 18(2):332–343
Weir JT, Schluter D (2008) Calibrating the avian molecular clock. Mol Ecol 17(10):2321–2328
Whitney BM, Vasconcelos MF, Silveira LF, Pacheco JF (2010) Scytalopus petrophilus (Rock Tapaculo): a new species from Minas Gerais, Brazil. Rev Bras Ornitol 18:73–88
Zeng K, Shi S, Wut CI (2007) Compound tests for the detection of hitchhiking under positive selection. Mol Biol Evol 24(8):1898–1908
Acknowledgments
We thank R.G. Lima, A. Martensen, M. Marini, A. Uezu and J.P. Metzger for providing us with some tissue samples. We also thank IBAMA, Instituto Florestal (SP, Brazil), Instituto Estadual de Florestas (MG, Brazil) and the Ministerio de Ecología de Misiones (Argentina) for the appropriate permits. We are also grateful to Conservación Argentina (G. Zurita and D. Varela), J. Albuquerque, F. d′Horta, R. Pessoa, F. Nodari and T. Matsumoto for assistance during fieldwork. V.A. Ellis, L. Calderon and L. Campagna provided useful comments that improved an early version of the manuscript. Finally, we thank J. Fjeldså and the Associate Editor F. Bairlein for the revision and comments that helped to improve the manuscript. This study was funded by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Brazil), Consejo Nacional de Investigaciones Científicas y Técnicas (Argentina, grant PIP 276), Agencia Nacional de Promoción Científica y Tecnológica (Argentina, grant PICT 805), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Brazil), Fundação de Amparo à Pesquisa do Estado de São Paulo (Brazil) and WWF (USA). All experiments comply with the current laws of the country in which they were performed. The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Fjeldså.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Cabanne, G.S., Sari, E.H.R., Meyer, D. et al. Matrilineal evidence for demographic expansion, low diversity and lack of phylogeographic structure in the Atlantic forest endemic Greenish Schiffornis Schiffornis virescens (Aves: Tityridae). J Ornithol 154, 371–384 (2013). https://doi.org/10.1007/s10336-012-0901-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10336-012-0901-8