
Abstract
The Japanese macaques (Macaca fuscata yakui) on Yakushima Island are an endemic subspecies and are closely related to the population of Kyushu, one of the main islands of Japan. Using feces collected throughout Yakushima Island, we examined mitochondrial DNA (mtDNA) to investigate the phylogeography of Japanese macaques. Six haplotypes were observed for a 203-bp fragment of the mtDNA control region. The nucleotide diversity (π) was low (0.0021). The genetic divergence within the Yakushima population was lower (0.009) than that among four haplotypes of the Kyushu population (0.015), calculated using Kimura’s two-parameter method. The mismatch distribution analysis of the six haplotypes of the Yakushima population suggested that the Yakushima population had experienced a sudden expansion in population size, which could be related to the bottleneck effect. The geographic distribution of the mtDNA haplotypes was not uniform. One haplotype was distributed widely, whereas the other five haplotypes were distributed only in the lowlands. The low genetic diversity and biased distribution are discussed in relation to an environmental crash caused by ancient volcanic activity near this island, which is postulated to have happened about 7,300 years ago, and the delayed recovery of highland vegetation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Various genetic markers have been used to clarify phylogeographic distribution patterns and to reveal genetic structure among populations since the work by Avise et al. (1987). These studies have surveyed huge areas, such as Eurasia or North America, as the habitats of several species. In Japan, several papers have reported the phylogeographic distribution and genetic structures of native terrestrial mammals, including the Japanese macaque (Macaca fuscata; Nozawa et al. 1991), brown bear (Ursus arctos yesoensis; Matsuhashi et al. 1999), sika deer (Cervus nippon; Goodman et al. 2001), and Eurasian badger (Meles meles; Kurose et al. 2001). These studies have revealed that terrestrial mammals in Japan were influenced by environmental changes during the Pleistocene, such as the formation of a land bridge between Asia and the Japanese archipelago. The progenitors of Japanese macaques have been distributed in the Japanese archipelago since the early part of the late Pleistocene (Kawamura 1994), inferred from the two fossil teeth (Iwamoto and Hasegawa 1972). However, it is difficult to discuss the environment during the dispersal of these animals after their arrival on the Japanese archipelago. For Japanese macaques, Kawamoto (1999) found that the geographic distribution of mtDNA haplotypes was homogeneous over a large area of northeastern Japan. Referring to the paleovegetation map of the Japanese archipelago (Tukada 1982), he suggested postglacial expansion of ancestors in the northeastern part of their current habitats. During the glacial period, the subarctic coniferous forest was unsuitable habitat for extant macaques. The rapid recovery of vegetation such as beech (Fagus crenata; Tomaru et al. 1997) may have enabled macaques to expand rapidly in that area. These results imply that the distribution of macaques is influenced not only by contiguous land, but also by climate or vegetation changes. The geographic distribution of animals depends on changes in food resources, such as vegetation types. Uehara (1975) pointed out the similarity of the present vegetation of the Japanese archipelago and the Korean peninsula and hypothesized that the Japanese macaque was able to shift its habitat from Korea to Japan via the land bridge at the marine isotope stage (MIS) 12 (0.43–0.48 Ma). Based on this postulate, we examined the geographic distribution of mtDNA haplotypes in the Japanese macaque (M. f. yakui) on Yakushima Island (hereinafter called Yakushima macaque), where the history of vegetation fluctuations in several altitudinal zones is partially known.
Materials and methods
Study area
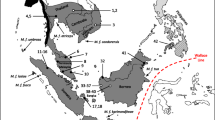
Yakushima Island is located in the temperate zone of southwestern Japan (30.3°N, 130.5°E; 0–1,936 m asl; Fig. 1). The island has an area of 504.9 km2 (Geographical Survey Institute, Japan 2004). The mean annual rainfall ranges from 2,926.2 mm (from May 2001 to April 2002; R. Tujino, unpublished data) to 4,358.7 mm [from 1971 to 2000; Japan Meteorological Agency (JMA) 2004] in the coastal area, and it exceeds 8,600 mm in the highlands (Eguchi 1984). In the coastal area, the mean annual temperature is 19.2°C, and the mean temperature of the coldest month is 8.4°C in January (from 1971 to 2000; data from JMA web page). The flora of this island is diverse owing to the warm Japanese Current, ample rainfall, and very steep topography.

Map of the five sampling sites of Japanese macaques (Macaca fuscata). The shaded island in the left map is Kyushu Island
Study population
The Yakushima macaque is distributed widely on the island, from subtropical broadleaf forest through cool temperate coniferous forest to subalpine grassland (Yoshihiro et al. 1999). The population density ranges from 1.3 to 2.4 km−2 in response to the production of fruit in each altitudinal zone (Hanya et al. 2004). Because mtDNA is inherited matrilineally (Giles et al. 1980) and females remain in their natal groups while males migrate to other groups after reaching sexual maturity (Sprague et al. 1998; Itani 1972), specific mtDNA haplotypes are characteristic of specific geographic regions. Details of the study site and the Yakushima macaque of the western coastal area of this island were described by Yamagiwa and Hill (1998).
Sampling monkey feces and laboratory methods
We collected 488 fecal samples between 1999 and 2002, including 91 samples reported in our previous study (Hayaishi and Kawamoto 2002). We reconnoitered mountain paths, forest roads, and local streets searching for fresh feces. To avoid resampling the same individual, each dropping was distinguished by freshness, size, shape, and color, and feces found less than 1.5 m apart were not sampled. The fecal samples were not used for genetic sexing, so they represented both sexes at various ages.
We collected epithelial cells, which had been cast off from the intestinal wall, from the fecal surface with a cotton swab, and dipped the swab in 2 ml of lysis buffer (White and Densmore 1992). The lysis buffer contained 0.5% SDS, 100 mM EDTA (pH 8.0), 100 mM Tris-HCl (pH 8.0), and 10 mM NaCl. The samples were stored in lysis buffer at room temperature. We extracted DNA from the fecal samples using the method of Kan et al. (1977), with the following modifications. Samples were incubated at 37°C for 2 h, adding 40 μl of proteinase K (10 mg/ml), 500 μl of salted TE, and 200 μl of 10% SDS. The digested protein was then removed by phenol–chloroform treatment for 2 h at room temperature on a rotating wheel. After centrifugation (10 min, 3,000 rpm), the supernatant was packed in dialysis membrane, and dialyzed overnight in 2 l of 20 μM Tris–HCl and 2 μM EDTA. Afterwards, the samples were stored with a drop of chloroform in vials at −20°C before PCR analysis.
We also used four individual samples from female Japanese macaques from four locations on Kyushu: Takasakiyama, Koshima, Kanoya, and Kushima (Fig. 1). Genomic DNA was extracted from blood using the method described above and was stored at the Primate Research Institute, Kyoto University.
To amplify 203 nucleotides of the second hypervariable segment of the D-loop region, we conducted nested PCR with two pairs of primers. The primers used for the first PCR were Saru4 and Saru5 (forward, 5′-ATCAGGGTCTATCACCCTAT-3′ and reverse, 5′-GGCCAGGACCAAGCCTATTTG-3′: Hayasaka et al. 1991), and the second internal PCR primers were mdl99 and mdl341 (forward, 5′-TATGCTGACTCCCACCACAT-3′ and reverse 5′-GTTTGGATGAAGGTCGGAGA-3′: Mouri et al. 2000).
Amplification took place in reaction volumes of 12.5 μl (containing 1.5 μl of template DNA) for the first PCR and 25 μl (containing 3 μl of the first PCR product) for the second PCR. Taq DNA polymerase (TaKaRa Shuzo) was used for one cycle at 94°C for 5 min, followed by 40 cycles at 94°C for 0.5 min, 48°C for 0.5 min, and 72°C for 0.5 min, with a final extension at 72°C for 5 min. A negative control was run with each PCR reaction to detect contamination. The amplified product of the second PCR was electrophoresed in a 3% low-melting-temperature agarose gel (NuSieve GTG agarose), then excised from the gel. When no band was visible, the PCR template was cleaned up before amplification with the Wizard SV Gel and PCR Clean-Up System (Promega), concentrated 10 times, and subjected to PCR again.
Direct sequencing was carried out with an ABI PRISM 310 or ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). The two internal primers, mdl99 and mdl341, were used as sequencing primers.
Data analysis
A multiple sequence alignment was constructed using CLUSTALX (Jeanmougin et al. 1998). Genetic distances were estimated using Kimura’s (1980) two-parameter distance method and the proportion (p)-distance method as implemented in MEGA version 2.1 (Kumar et al. 2001). The indels and transversions were given the same weight as transitions in estimating genetic distance. Phylogenetic trees were reconstructed using the neighbor-joining method (Saitou and Nei 1987) using MEGA. A parsimonious network was constructed using TCS version 1.13 (Clement et al. 2000). Nucleotide diversity (π; Nei 1987) was estimated using ARLEQUIN version 2.000 (Schneider et al. 2000). The haplotype diversity (ĥ) was calculated for all specimens and by altitudinal zone (see below) using the formula \( \hat{h} = 2n(1 - \sum {\hat{x} } _i^2 )/(2n - 1), \) where n is sample number, and \( \hat{x} _i \) is each haplotype frequency (Nei 1987). The expected value of ĥ was calculated with the haplotype frequency of all specimens. The expected pairwise mismatch distribution under models of population expansion or equilibrium (Rogers and Harpending 1992; Rogers 1995) was calculated and the goodness of fit was tested using ARLEQUIN. The mismatch analysis suggests the effects of past demography. When a sudden population expansion occurs, the distribution is expected to become unimodal. When the population grows without demographic fluctuation, the form becomes multimodal.
Results
Sequence analyses, phylogenetic analyses
We collected 488 fecal samples and used them for PCR amplification. The success rate of amplification with the nested primers and sequence analyses was 57.4%. Of the 488 amplified fecal samples, 280 were successfully sequenced. Sequencing of 203-bp of the mitochondrial DNA control region for 280 samples of Yakushima macaques yielded a total of six unique haplotypes. We identified five variable sites: four transitions and one transversion. The haplotypes were named Yakushima1 to Yakushima6 (abbreviated Y1 to Y6, respectively). We have previously reported four haplotypes: Y1–Y4 (Hayaishi and Kawamoto 2002). The base substitution sites with reference to the sequence of M. mulatta obtained from DDBJ (DNA Data Bank of Japan), accession no. AY612638, sample size and haplotype frequencies are shown in Table 1. The mean nucleotide diversity (π) was 0.002 in the Yakushima macaque. In addition, three haplotypes of Japanese macaque (M. f. fuscata) were determined from the four areas, Takasakiyama (JN43), Koshima (JN44), Kanoya (JN44), and Kushima(JN45) in Kyushu (the codes in parentheses were names of haplotypes distinguished with the 412-bp sequence of mtDNA control region, by Y. Kawamoto et al., submitted). The obtained mtDNA sequences of the Yakushima macaque are available from DDBJ under accession numbers AB093529-AB093532, AB181509 and AB181510.
The mean pairwise nucleotide distance was 0.009 between the Yakushima haplotypes, 0.015 between the Kyushu haplotypes, and 0.047 between the Yakushima and Kyushu haplotypes. A phylogenetic tree was constructed using the neighbor-joining method. The six Yakushima haplotypes clustered into one group and near southern Kyushu (Koshima, Kushima and Kanoya) haplotypes with M. mulatta as the outgroup (Fig. 2).

A control region neighbor-joining tree for six haplotypes of M. f. yakui and four haplotypes of M. f. fuscata in Kyushu with M. mulatta as the outgroup. Numbers of nucleotide substitution per site indicated by the scale are the Kimura’s (1980) two-parameter distances. The numbers on each branch indicated the bootstrap value (>70) after 1,000 time replications
The six Yakushima haplotypes were connected using the program TCS. A parsimonious haplotype network revealed a star-shaped phylogeny for the mtDNA variation (Fig. 3).

The star-like phylogenetic network represents the haplotype relationships of M. f. yakui. Each line between haplotypes was one mutation step. The size of circles indicates the haplotype frequency. TCS software was used in this analysis
Mismatch distribution
The distribution of pairwise differences was constructed to test the hypothesis of population expansion in the Yakushima macaque. The mismatch distribution fit the expected distribution under a model of population expansion well (SSD=0.0012, P=0.53, and r=0.26, P=0.62). The raggedness index r (Harpending 1994) indicated a smooth distribution, reflecting a model of sudden expansion (Fig. 4). The unimodal distribution is observed when a sudden expansion was occurred in the population. In addition to the low genetic diversity of Yakushima macaques, the differences of bases was very small in Yakushima macaques (Fig. 4) suggesting the sudden expansion after the population bottleneck.

Mismatch distribution established for M. f. yakui. The thin line represents the observed mismatch distribution. The dotted line represents the expected mismatch distribution
Haplotype distribution
The geographic distribution of the six haplotypes was not uniform on the island. Y1 was distributed widely, while the other five haplotypes were found at lower altitudes (Fig. 5). The altitudinal distribution of six haplotypes were significantly biased, tested Fisher’s exact test (χ²=25.62, df=15, P=0.042; Table 2). The haplotype diversity (ĥ) differed among altitudinal zones defined after Hanya et al. (2004) (0–399 m: n=176, ĥ=0.364; 400–799 m: n=57, ĥ=0.326; 800–1,199 m: n=30, ĥ=0; 1,200–1,699 m: n=17, ĥ=0). The haplotype diversity of all 280 specimens was 0.305, so the specimens from the two lower altitudinal zones have contributed much more to the increased haplotype diversity than those from the two higher altitudinal zones.

Spatial distributions of mtDNA haplotypes of M. f. yakui in Yakushima Island. The bar at the bottom indicated distance of 10 km. Six symbols indicated Y1 (open circle), Y2 (closed circle), Y3 (open rectangle), Y4 (open diamond), Y5 (closed star) and Y6 (closed hexagon), respectively. The outline indicated a shoreline, and contours were drawn at 400, 800, 1,200, 1,700 m asl
Discussion
Genetic diversity
The six haplotypes of Yakushima macaque were considered specific to the Yakushima population, as these haplotypes were unique to the Yakushima macaque and formed a single cluster separated from the Kyushu population (Fig. 2). Mouri et al. (2000) reported that out of several populations of M. fuscata, only the Yakushima macaque had a one-base insertion in the mtDNA control region, which we confirmed in this study (Table 1). This suggests that the Yakushima population of Japanese macaques is unique in terms of their mitochondrial DNA. The low genetic differentiation in the mtDNA of Yakushima macaques was confirmed, which concurs with previous findings of blood protein polymorphism (Shotake et al. 1975; Nozawa et al. 1977, 1982, 1991).
Demographic history
Yakushima Island was connected with Kyushu by a land bridge when sea levels dropped during several past glacial stages (Kimura 2000). The Yakushima macaques could have migrated via those land bridges. The estimated divergence time between the Yakushima and Kyushu populations is 178 ka, as calculated from the genetic distance using a blood protein (Nozawa et al. 1977). Nozawa et al. (1977) assumed that gene flow from Kyushu to Yakushima after the MIS 6–8 (0.13–0.30 Ma) should be very rare or unlikely, even if a land bridge formed during the MIS 2–4 (10–80 ka). Our result of the different sequences of mtDNA supported this assumption that female monkeys did not migrate between Kyushu and Yakushima.
From the viewpoint of an island population (Frankham 1997), genetic diversity should be lower on Yakushima than in the Kyushu population. Generally, an island population has less space for refugia than a mainland population during natural calamities, so the effect of a population bottleneck is more significant.
The star-shaped phylogeny (Fig. 3) suggests that the Yakushima population has undergone population expansion at least once. The results of the mismatch analysis also suggest that the Yakushima population has experienced a bottleneck event followed by population expansion (Fig. 4). We observed only 1 or 2 base substitutions among the six haplotypes. This small difference is unlikely to have resulted from a founder effect that occurred as long as 130–200 ka. Some geological events have been reported in the South Kyushu area, including Yakushima Island, after the last glacial period (from ca. 10 ka until today; Machida and Arai 1992). A great explosive eruption from the Kikai caldera (Machida and Arai 1978) ca. 7,300 calendar years ago (Fukuzawa 1995), which produced the Koya pyroclastic flow (Ui 1973), was the most recent explosion near Yakushima, and it occurred 40 km northwest of the island. The Kikai caldera explosion likely caused the extinction of some mammals on Yakushima Island and decreased the genetic diversity of the surviving animals. The result suggests the occurrence of a recent population bottleneck involving Yakushima macaques. Therefore, it is necessary to determine when the bottleneck affected the Yakushima macaque population. Additional mtDNA sequence data for the Kyushu macaque population are needed to estimate the mutation rate (μ) of the control region correctly. The time passed from the expansion (T) will be estimated with the expansion value τ , calculated by ARLEQUIN (Schneider et al. 2000), using the formula τ=2μT (Rogers and Harpending 1992).
The unimodal mismatch distribution indicates a recent population expansion in the Yakushima macaque (Fig. 4). The low level of DNA diversity also suggests that the Yakushima population has experienced a population bottleneck.
Geographically biased distribution
The geographic distribution of the six haplotypes was not uniform (Fig. 5), although the 280 specimens were collected evenly in an attempt to fit the distribution of monkeys according to the group density reported by Yoshihiro et al. (1998, 1999). Y1 was distributed widely, as expected from the estimated frequency, whereas the other five haplotypes were observed only in restricted areas in the lowland forests. This suggests that the geographically biased distribution of the six haplotypes was influenced by heterogeneity of the recovery history of the vegetation across the altitudinal zones. The total annual fruit production influences the population density of monkeys inhabiting three altitudinal zones (Hanya et al. 2004). Therefore, theoretically, environmental heterogeneity would influence the dispersion of animals (Shigesada et al. 1986).
The distributions of some haplotypes in Fig. 5 (e.g., Y3 and Y5) were highly restricted. This result was probably be influenced by the female philopatry of Japanese macaques and the matrilineal splitting pattern of troops (Oi 1988). The geographical pattern also suggest that new troops after the troop fission have ranged near by the previous home range in lowland forests.
Our results suggest that the altitudinal zones and vegetation fluctuations influenced the geographic distribution of mtDNA haplotypes. The geographically biased distribution is likely influenced by the geological history of Yakushima Island and historical differences in the recovery of the plant community with reference to archaeobotanic studies (S. Hayaishi et al., in preparation). All of the non-Y1 haplotypes would be derived from Y1 (Fig. 3) and probably evolved during the population expansion at a time during the vegetation recovery in the lowland forests where the recovery was most advanced. Uehara (1975) discussed the importance of the woody plants of the cool temperate forest to the adaptation of Japanese macaques to the cool temperate zone. Our study also suggests that Japanese macaques have an adaptive advantage in the warm temperate forest over the cool temperate forest on Yakushima Island, which partly supports his hypothesis.
References
Avise JC, Arnold J, Ball RM, Berminghame E, Lamb T, Neigel JE, Reeb CA, Saunders NC (1987) Intraspecific phylogeography—the mitochondrial-DNA bridge between population-genetics and systematics. Annu Rev Ecol Syst 18:489–522
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Eguchi T (1984) Climate of Yaku-shima Island, especially regionality of precipitation distribution. In: Nature Conservation Bureau, Environment Agency, Japan (eds) Conservation reports of the Yaku-shima wilderness area, (in Japanese with English summary). Kyushu, Japan, pp 3–26
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity 78:311–327
Fukuzawa H (1995) Non-glacial varved lake sediment as a natural timekeeper and detector on environmental changes (in Japanese with English abstract). Quat Res 34:135–149
Geographical Survey Institute, Japan (2004) http://www.gsi.go.jp/
Giles RE, Blanc H, Cann HM, Wallace DC (1980) Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci USA 77:6715–6719
Goodman SJ, Tamate HB, Wilson R, Nagata J, Tatsuzawa S, Swanson GM, Pemberton JM, Mccullough DR (2001) Bottlenecks, drift and differentiation: the population structure and demographic history of sika deer (Cervus nippon) in the Japanese archipelago. Mol Ecol 10:1357–1370
Hanya G, Yoshihiro S, Zamma K, Matsubara H, Ohtake M, Kubo R, Noma N, Agetsuma N, Takahata Y (2004) Environmental determinants of the altitudinal variations in relative group densities of the Japanese macaques on Yakushima. Ecol Res 19:485–493
Harpending RC (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol 66: 591–600
Hayaishi S, Kawamoto Y (2002) Fecal genotyping of mitochondrial DNA polymorphism and fecal age-class estimation of Macaca fuscata yakui: a preliminary report (in Japanese with English abstract). Honyurui Kagaku 42:161–166
Hayasaka K, Ishida T, Horai S (1991) Heteroplasmy and polymorphism in the major non-coding region of mitochondrial DNA in Japanese monkeys: association with tandemly repeated sequences. Mol Biol Evol 8:399–415
Itani J (1972) Social structure of primates (in Japanese). Kyoritsu Shuppan, Tokyo
Iwamoto M, Hasegawa Y (1972) Two macaque fossil teeth from the Japanese Pleistocene. Primates 13:77–81
Japan Meteorological Agency (2004) http://www.jma.go.jp/
Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ (1998) Multiple sequence alignment with Clustal X. Trends Biochem Sci 23:403–405. DOI 10.1016/S0968-0004(98)01285-7
Kan YW, Dozy AM, Trecartin R, Todd D (1977) Identification of a nondeletion defect in α-thalassemia. N Engl J Med 297:1081–1084
Kawamura Y (1994) Late Pleistocene to Holocene mammalian faunal succession in the Japanese islands, with comments on the late Quaternary extinctions. Archaeozoologia 6:7–22
Kawamoto Y (1999) Origin of Japanese macaques in views from genes (in Japanese). Kagaku 69:300–305
Kimura M (1980) A simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kimura M (2000) Paleogeography of the Ryukyu Islands. Tropics 10:5–24
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244–1245
Kurose N, Kaneko Y, Abramov AV, Siriaroonrat B, Masuda R (2001) Low genetic diversity in Japanese population of the Eurasian badger Meles meles (Mustelidae, Carnivora) revealed by mitochondrial cytochrome b gene sequences. Zool Sci 18:1145–1151
Machida H, Arai F (1978) Akahoya Ash-A Holocene widespread tephra erupted from the Kikai Caldera, south Kyushu, Japan (in Japanese with English abstract). Quat Res 17:143–163
Machida H, Arai F (1992) Atlas of tephra in and around Japan (in Japanese). University of Tokyo Press, Tokyo
Matsuhashi T, Masuda R, Mano T, Yoshida MC (1999) Microevolution of the mitochondrial DNA control region in the Japanese brown bear (Ursus arctos) population. Mol Biol Evol 16:676–684
Mouri T, Agatsuma T, Iwagami M, Kawamoto Y (2000) Species identification by mitochondrial DNA: a case study of macaque remains from Shuri Castle, Okinawa (in Japanese with English abstract). Primate Research 16:87–94
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nozawa K, Shotake T, Ohkura Y, Tanabe Y (1977) Genetic variations within and between species of Asian macaques. Jpn J Genet 52:15–30
Nozawa K, Shotake T, Kawamoto Y, Tanabe Y (1982) Population genetics of Japanese monkeys: II. Blood protein polymorphisms and population structure. Primates 23:252–271
Nozawa K, Shotake T, Minezawa M, Kawamoto Y, Hayasaka K, Kawamoto S, Ito S (1991) Population genetics of Japanese monkeys: III Ancestry and differentiation of local populations. Primates 32:411–435
Oi T (1988) Sociological study on the troop fission of wild Japanese monkeys (Macaca fuscata yakui) on Yakushima Island. Primates 29:1–19
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Rogers AR (1995) Genetic evidence for a Pleistocene population explosion. Evolution 49:608–615
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schneider S, Roessli D, Excoffier L (2000) Arlequin: a software for population genetics data analysis. Ver 2.000. Genetics and Biometry Lab, Department of Anthropology, University of Geneva
Shigesada N, Kawasaki K, Teramoto E (1986) Traveling periodic waves in heterogeneous environments. Theor Popul Biol 30:143–160
Shotake T, Ohkura Y, Nozawa K (1975) A fixed state of the PGM-2mac allele in the population of the Yaku macaque (Macaca fuscata yakui). In: Contemporary Primatology, 5th International Congress on Primates, Nagoya 1974. Karger, Basel, pp 67–74
Sprague DS, Suzuki S, Takahashi H, Sato S (1998) Male life history in natural populations of Japanese macaques: migration, dominance rank, and troop participation of males in two habitats. Primates 39:351–363
Tomaru N, Mitsutsuji T, Takahashi M, Tsumura Y, Uchida K, Ohba K (1997) Genetic diversity in Fagus crenata (Japanese beech): influence of the distributional shift during the late-Quaternary. Heredity 78:241–251
Tukada M (1982) Cryptomeria japonica: glacial refugia and late-glacial and postglacial migration. Ecology 63:1091–1105
Uehara S (1975) The importance of the temperate forest elements among woody food plants utilized by Japanese monkeys and its possible historical meaning for the establishment of the monkeys’ range—a preliminary report. In: Contemporary Primatology, 5th International Congress on Primates, Nagoya 1974. Karger, Basel, pp 392–400
Ui T (1973) Exceptionally far-reaching, thin pyroclastic flow in southern Kyushu, Japan (in Japanese with English abstract). Bull Volc Soc Jpn, Second Ser 18:153–168
White OL, Densmore III LD (1992) Mitochondrial DNA isolation. In: Hoelzel AR (eds) Molecular genetic analysis of populations, a practical approach, 1st edn. IRL Press, Oxford, pp 29–55
Yamagiwa J, Hill DA (1998) Intraspecific variation in the social organization of Japanese macaques: past and present scope of field studies in natural habitats. Primates 39:257–273
Yoshihiro S, Furuichi T, Manda M, Ohkubo N, Kinoshita M, Agetsuma N, Azuma S, Matsubara H, Sugiura H, Hill D, Kido E, Kubo R, Matsushima K, Nakajima K, Maruhashi T, Oi T, Sprague D, Tanaka T, Tsukahara T, Takahata Y (1998) The distribution of wild Yakushima macaque (Macaca fuscata yakui) troops around the coast of Yakushima Island, Japan. Primate Research 14:179–187
Yoshihiro S, Ohtake M, Matsubara H, Zamma K, Hanya G, Tanimura Y, Kubota H, Kubo R, Arakane T, Hirata T, Furukawa M, Sato A, Takahata Y (1999) Vertical distribution of wild Japanese macaques (Macaca fuscata yakui) in the western area of Yakushima Island, Japan: preliminary report. Primates 40:409–415
Acknowledgements
We thank Drs. T. Nishida, T. Shotake, O. Takenaka, J. Yamagiwa, T. Hikida, S. Suzuki, members of Laboratory of Human Evolution Studies and Dr. D.G. Smith, two anonymous reviewers and Editor-in-chief for fruitful and constructive comments on this paper. And we thank Mr. R. Tsujino for using unpublished data and some participants of Yakushima Macaque Research Group for helping us collecting fecal samples; Drs. Y. Tanaka, A. Yamane, T. Hikida, T. Sota and K. Inaba for helping us in the laboratory; Kamiyaku-cho, Yaku-cho and Yakushima District Forestry Office for permission to this study; Field Research Center of Primate Research Institute, Kyoto University for permission to use the field station in Yakushima and Dr. A. Satoh and many friends in Yakushima Island for their help and hospitality during our fieldwork. This study was financed by Cooperative Research Fund of Primate Research Institute, Kyoto University, and partly by grants-in-aid for Scientific Research, Ministry of Education, Culture, Sports, Science and Technology 09358017 to Dr. T. Shotake, 10CE2005 to Dr. O. Takenaka and 21st Century COE Research Kyoto University (A14).
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Hayaishi, S., Kawamoto, Y. Low genetic diversity and biased distribution of mitochondrial DNA haplotypes in the Japanese macaque (Macaca fuscata yakui) on Yakushima Island. Primates 47, 158–164 (2006). https://doi.org/10.1007/s10329-005-0169-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10329-005-0169-1