
Abstract
Organic reactions in aqueous media are being developed because water is environmentally benign. The Horner–Wadsworth–Emmons reaction is a modified Wittig reaction for the synthesis of α,β-unsaturated ketones and other conjugated compounds. Here we prepared high molecular weight ketones by the Horner–Wadsworth–Emmons reaction of dimethyl-2-oxopropylphosphonate and various aldehydes in water at room temperature. The product was precipitated during the reaction process and was separated readily by a simple filtration in 90–99 % yield.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Wittig reaction is an important method for the formation of new carbon–carbon bonds in synthetic organic chemistry and in natural products (Britton et al. 1996). One of the modified Wittig reactions is the use of the inexpensive and non-toxic dialkyl-2-oxopropylphosphonates in reactions involved in α,β-unsaturated compounds formation which was first introduced by Horner–Wadsworth–Emmons (Wadsworth 2005; Wadsworth and Emmons 1961; Horner et al. 1959; Sano et al. 2002; Ando 1998; Arai et al. 1998; Johnson and Zhang 1995; Blanchette et al. 1984). The Horner–Wadsworth–Emmons reaction is an important method for the syntheses of α,β-unsaturated ketones and other conjugated compounds. Other parallel methods for production of α,β-unsaturated systems such as Knoevenagel and aldol condensation have drawbacks such as multiplicity of procedures, side reactions, high temperature, low yield and low stereoselectivity (Choudary et al. 1999; Zumbansen et al. 2010).
Stereodefined synthesis of carbon–carbon double bonds with high selectivity is critically important in organic synthesis. The Wittig and related reactions have served as the most powerful method in the construction of double bonds. The Horner–Wadsworth–Emmons modification of the Wittig reaction is especially widely employed in pharmaceutical synthesis. In general, the Horner–Wadsworth–Emmons reaction preferentially gives stable E-α,β-unsaturated esters and ketones, and therefore, it can be a superior replacement for aldol condensation reactions (Roelfs et al. 2000; Tichit et al. 2002). However, various studies on Horner–Wadsworth–Emmons reaction indicate some limitation in reaction conditions such as the use of toxic solvents and very strong bases [lithium diisopropylamide, Grignard reagent, lithium bis(trimethylsilyl)amide, n-butyllithium, sodium hydride, potassium bis(trimethylsilyl)amide] or expensive catalyst, moderate yields and very low temperature to control stereoselectivity (Pihko and Salo 2003; Rathke and Nowak 1985; Sano et al. 2003).
Presently, organic reactions in aqueous media have attracted many researchers because water is a solvent with advantages of environmentally benign and economically affordable. As part of our ongoing program toward the development of green chemistry, we have conducted different organic reactions in water (Jafari and Ghadami 2015; Jafari and Mahmoudi 2013; Jafari et al. 2009a, b, 2012; Ghadami and Jafari 2015). Therefore in this paper the Horner–Wadsworth–Emmons reaction was performed in water by available, inexpensive NaOH as base, non-toxic dimethyl-2-oxopropylphosphonate and different aldehydes at room temperature. Under this environmentally friendly conditions, an excellent chemo- and trans-selectivity was observed in the Horner–Wadsworth–Emmons reactions of dimethyl-2-oxopropylphosphonate and different aromatic aldehydes.
Experimental
General
Melting points were determined with a Buchi melting point 540 apparatus and were uncorrected. 1HNMR (400 MHz) and 13CNMR (100 MHz) spectra were recorded on a Bruker Advance 400 spectrometer in DMSO (d6) using tetramethylsilane as internal reference. All chemicals were purchased from Aldrich, Merck and Fluka. Distilled water was used for all reactions.
General procedure
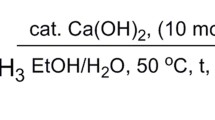
Aromatic aldehyde (1 mmol) was added to the test tube containing NaOH (4 mL, 1 M) and dimethyl-2-oxopropylphosphonate (1.1 mmol, 0.182 g), while the reaction mixture was stirred and the stirring was continued at room temperature for the prescribed time (Table 2) until the reaction was completed (thin-layer chromatography). Then, the ice water was added to the reaction mixture, and the solid product was simply filtered off and washed with cold water. The sole product was obtained after drying at room temperature in high to excellent yield.
Selected spectral data
4-(4-Nitrophenyl)but-3-en-2-one (Table 2, entry 1): trans/cis isomer (100.0/0.0); yellow solid; melting point 116–117 °C; IR: υ = 1691, 1668, 1594, 1509, 1419, 1335, 1255, 1182, 1110, 974, 860, 825, 746 cm−1; 1H NMR (400 MHz, CDCl3) δ = 8.29 (d, J = 8.8 Hz, 2H), 7.72 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 16.4 Hz, 1H), 6.83 (d, J = 16.4 Hz, 1H), 2.45 (s, 3H).
4-Phenyl-but-3-en-2-one (Table 2, entry 2): trans/cis isomer (100.0/0.0); bright yellow solid; melting point: 41–42 °C; IR: υ = 1686, 1599, 1452, 1361, 1315, 1177, 982, 908, 750, 693 cm−1; 1H NMR (400 MHz, CDCl3) δ = 7.56–7.53 (m, 2H), 7.52 (d, J = 16.4 Hz, 1H), 7.40–7.38 (m, 3H), 6.72 (d, J = 16.4 Hz, 1H), 2.38 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ = 198.5, 143.5, 130.5, 130.0, 129.0, 128.3, 127.1, 27.5.
4-(3-Hydroxyphenyl)but-3-en-2-one (Table 2, entry 3): trans/cis isomer (100.0/0.0); bright yellow; melting point 96–97 °C; IR: υ = 3500–2500 (br, OH), 3075, 1624, 1576, 1489, 1362, 1249, 961, 777 cm−1; 1H NMR (400 MHz, CDCl3) δ = 7.51 (d, J = 16.4 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.21 (bs, OH, 1H), 7.11 (s, 1H), 7.10 (d, J = 8.0 Hz, 1H), 6.97 (d, J = 8.0, 1.2 Hz, 1H), 6.71 (d, J = 16.4 Hz, 1H), 2.42 (s, 3H).
4-(4-Chloro-phenyl)-but-3-en-2-one (Table 2, entry 4): trans/cis isomer (100.0/0.0); white solid; melting point 105–106 °C; IR: υ = 1657, 1588, 1488, 1406, 1359, 1201, 1089, 1010, 976 cm−1; 1H NMR (400 MHz, CDCl3) δ = 7.48 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 16.0 Hz, 1H), 7.37 (d, J = 8.4 Hz, 2H), 6.69 (d, J = 16.0 Hz, 1H), 2.38 (s, 3H); 13C NMR (100 MHz, CDCl3) δ = 196.1, 141.8, 136.4, 132.9, 129.4, 129.2, 127.5, 27.7.
6-Phenyl-hex-3,5-dien-2-one (Table 2, entry 5): trans/cis isomer (100.0/0.0); White solid; melting point 55–56 °C; IR: υ = 1651, 1615, 1449, 1363, 1294, 1253, 1149, 1074, 982, 819, 749, 692 cm−1; 1H NMR (400 MHz, CDCl3) δ = 7.26–7.48 (m, 6H), 6.89–6.98 (m, 2H), 6.26 (d, J = 15.6 Hz, 1H), 2.31 (s, 3H); 13C NMR (100 MHz, CDCl3) δ = 198.8, 143.7, 141.4, 135.9, 130.4, 129.3, 128.9, 128.6, 128.5, 127.2, 126.6, 27.3.
4-(4-Methylphenyl)but-3-en-2-one (Table 2, entry 6): trans/cis isomer (100.0/0.0); white solid; melting point 32–33 °C; IR: υ = 1664, 1607, 1513, 1255, 1177, 976, 801 cm−1; 1H NMR (400 MHz, CDCl3) δ = 7.51 (d, J = 16.4 Hz, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 6.70 (d, J = 16.4 Hz, 1H), 2.40 (s, 3H), 2.39 (s, 3H).
4-(4-Methoxyphenyl)-but-3-en-2-one (Table 2, entry 7): trans/cis isomer (100.0/0.0); white solid; melting point 72–73 °C; IR: υ = 1632, 1599, 1510, 1423, 1241, 1175, 971, 764 cm−1; 1H NMR (400 MHz, CDCl3) δ = 7.62 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 16.4 Hz, 1H), 6.94 (d, J = 8.8 Hz, 2H), 6.63 (d, J = 16.4 Hz, 1H), 3.88 (s, 3H), 2.38 (s, 3H).
4-(3-Nitro-phenyl)-but-3-en-2-one (Table 2, entry 8): trans/cis isomer (100.0/0.0); white solid; melting point 102–103 °C; IR: υ = 1672, 1616, 1522, 1530, 1264 cm−1; 1H NMR (400 MHz, CDCl3) δ = 8.41 (s, 1H), 8.25 (d, J = 7.6 Hz, 1H), 7.85 (d, J = 7.3 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 16.0 Hz, 1H), 6.84 (d, J = 16.0 Hz, 1H), 2.42 (s, 3H); 13C NMR (100 MHz, CDCl3) δ = 197.5, 140.1, 136.3, 133.7, 130, 129.4, 124.7, 122.6, 28.1.
4-(3,4-Dimethoxyphenyl)but-3-en-2-one (Table 2, entry 9): trans/cis isomer (100.0/0.0); white solid; melting point 59–60 °C; IR: υ = 1667, 1620, 1595, 1507, 1422, 1364, 1251, 1161, 1137, 1019, 977, 879, 805, 744 cm−1; 1H NMR (400 MHz, CDCl3) δ = 7.48 (d, J = 16.4 Hz, 1H), 7.15 (d, J = 8.0, 1.6 Hz, 1H), 7.09 (d, J = 1.6 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 6.62 (d, J = 16.4 Hz, 1H), 3.97 (s, 3H), 3.94 (s, 3H), 2.39 (s, 3H).
4-(4-(Dimethylamino)phenyl)but-3-en-2-one (Table 2, entry 10): trans/cis isomer (100.0/0.0); yellow solid; melting point 96–97 °C; IR: υ = 1673, 1575, 1356, 1164, 965, 804 cm−1; 1H NMR (400 MHz, CDCl3) δ = 7.48 (d, J = 16.4 Hz, 1H), 7.46 (d, J = 8.8 Hz, 2H), 6.69 (d, J = 8.8 Hz, 2H), 6.56 (d, J = 16.4 Hz, 1H), 3.05 (s, 6H), 2.36 (s, 3H).
Results and discussion
The conventional multistep preparation of a complex molecule generally involves a large number of synthetic operations, including extraction and purification processes. This leads to the synthetic inefficiency as well as generation of large amounts of waste. For instance, high molecular weight aldehydes and ketones are important chemicals usually prepared from condensation of short aldehydes or ketones in a three-step process consisting of a base-catalyzed aldol condensation, followed by the acid-catalyzed dehydration of the β-hydroxy aldehyde or β-hydroxy ketone and finally with a selective hydrogenation of the α,β-unsaturated aldehyde. In this procedure besides the desired reaction, several side reactions can take place such as self-condensations of aldehyde which is a potential problem (Roelfs et al. 2000). Therefore, the replacement of aldol condensation by a one-step Wittig reaction seems appropriate solution to this problem. In addition, it should be noted that recently, there has been an increasing concern in regard to the tight legislation on the maintenance of eco-friendly synthetic pathways and processes (Tundo et al. 2000). But, volatile organic solvents amount to over 85 % of the mass utilization in a typical chemical manufacturing process, and as the recovery efficiency is far from satisfactory, they are major contributors to the environmental pollution (Constable et al. 2007). Thus, to remove organic solvents from the chemical process, synthetic chemists are responsible for elimination of volatile organic solvents or their replacement with non-flammable, non-volatile, non-toxic and inexpensive solvents (Reinhardt et al. 2008). Therefore, one should select solvents that will have no or limited impact on health and the environment. As a reaction medium, water complies with all the current stringent requirements on sustainable chemistry. Consequently, in this paper water was selected as the reaction medium and the available, inexpensive NaOH was used as the catalyst. For optimization of the catalyst concentration, the reaction of benzaldehyde and dimethyl-2-oxopropylphosphonate was checked with different catalyst concentrations at room temperature.
Based on the results of Table 1, the reaction of benzaldehyde (1 mmol) and dimethyl-2-oxopropylphosphonate (1.1 mmol) in aqueous solution of NaOH (4 mL, 1 M) was selected as a model reaction at room temperature. The corresponding solid α,β-unsaturated ketone was obtained at 90 % isolated yield after a simple filtration and washing with water. Furthermore, under similar reaction conditions, a series of α,β-unsaturated ketones have been prepared by the use of different aldehydes in high to excellent yields (Table 2).

It was observed that both electron-rich and electron-deficient aromatic aldehydes reacted efficiently and formed trans-isomer of α,β-unsaturated ketones under this green condition. Excellent trans-selectivity of the reaction was proved by 1HNMR and melting point observations. In all cases, the reaction procedure was very simple and the desired solid product was easily separated by cooling, simple filtration, followed by washing with cold water and drying at room temperature. Furthermore, the reaction of acetophenone and cyclohexanone as ketone was also studied and found that no reaction occurred under similar reaction conditions.
The plausible role of water on excellent trans-selectivity and in promoting the reaction is rationalized by the mechanism described in Fig. 1. The hydrated phosphonate carbanion was easily formed by aqueous hydroxyl ion. Water molecule increased the electrophilic character at the carbonyl compound by hydrogen bond formation between water and the carbonyl oxygen atom of the aldehyde (Butler and Coyne 2010). These two planar groups are close to each other in minimal strict hindered to form oxaphosphetane intermediate. The trans-α,β-unsaturated ketones was produced after a pseudorotation, P–C bond together with O–C bond cleavage.

Proposed mechanism for the Horner–Wadsworth–Emmons reaction in water (the broken lines show hydrogen bondings): a Hydroxide ion converted dimethyl-2-oxopropylphosphonate to active phosphonate carbanion and hydrated, b aldehyde activated by hydrogen bonding, and c they are pushed near each other by hydrophobic effect to react more easily
Conclusion
We have developed a simple, safe and economic method for selective trans-olefination between dimethyl-2-oxopropylphosphonate and arylaldehydes. This procedure was performed for the first time in water. The multistep synthesis in toxic organic solvent and harsh chemical conditions was replaced with one-step reaction in water. In addition, high yields of the products, excellent chemo-selectivity, ease of workup and low cost make the above method advantageous in comparison with other existing methods.
References
Ando K (1998) Z-Selective Horner–Wadsworth–Emmons reaction of α-substituted ethyl (diarylphosphono)acetates with aldehydes. J Org Chem 63:8411–8416
Arai S, Hamaguchi S, Shioiri T (1998) Catalytic asymmetric Horner–Wadsworth–Emmons reaction under phase-transfer-catalyzed conditions. Tetrahedron Lett 39:2997–3000
Bhagat S, Sharma R, Chakraborti AK (2006) Dual-activation protocol for tandem cross-aldol condensation: an easy and highly efficient synthesis of α,α-bis(aryl/alkylmethylidene) ketones. J Mol Catal A: Chem 260(1–2):235–240
Blanchette MA, Choy W, Davis JT, Essenfeld AP, Masamune S, Roush WR, Sakai T (1984) Horner–Wadsworth–Emmons reaction: use of lithium chloride and an amine for base-sensitive compounds. Tetrahedron Lett 25(21):2183–2186
Britton G, Liaaen-Jensen S, Pfander, H (1996) Carotenoids volume 2: synthesis. Birkhäuser Verlag, Basel, pp 89–100
Butler RN, Coyne AG (2010) Water: nature’s reaction enforcers comparative effects for organic synthesis “In-Water” and “On-Water”. Chem Rev 110:6302–6337
Choudary BM, Kantam ML, Sreekanth P, Bandopadhyay T, Figueras F, Tuel A (1999) Knoevenagel and aldol condensations catalyzed by a new diamino-functionalized mesoporous material. J Mol Catal A Chem 142:361–365
Constable DJC, Gonzalez CJ, Henderson RK (2007) Perspective on solvent use in the pharmaceutical industry. Org Process Res Dev 11:v133–v137
Ghadami M, Jafari AA (2015) Efficient synthesis of Mannich bases by sonication in sodium dodecyl sulfate micellar media. Environ Chem Lett 13:191–196
Hayes WM (2013) CRC handbook of chemistry and physics. CRC Press, London
Horner L, Hoffmann H, Wippel HG, Klahre G (1959) Phosphororganische Verbindungen. XX. Phosphinoxydeals Olefin 92(10):2499–2505
Jafari AA, Ghadami M (2015) High yield room temperature synthesis of pyranochromenes in neutral cetyltrimethylammonium bromide micellar media. Environ Chem Lett. doi:10.1007/s10311-015-0546-y
Jafari AA, Mahmoudi H (2013) Room temperature aqueous Paal–Knorr pyrrole synthesis catalyzed by aluminum tris(dodecyl sulfate)trihydrate. Environ Chem Lett 11:157–162
Jafari AA, Moradgholi F, Tamaddon F (2009a) A highly efficient Michael addition of indoles to α,β-unsaturated electron deficient compounds in acidic SDS micellar media. J Iran Chem Soc 6:588–593
Jafari AA, Moradgholi F, Tamaddon F (2009b) Pronounced catalytic effect of a micellar solution of sodium dodecylsulfate (SDS) upon a three-component reaction of aldehydes, amines, and ketones under neutral conditions. Eur J Org Chem 8:1249–1255
Jafari AA, Amini S, Tamaddon F (2012) A green, chemoselective and efficient protocol for Pall–Knorr pyrrole and bispyrrole synthesis using biodegradable polymeric catalyst PEG–SO3H in water. J Appl Polym Sci 125:1339–1345
Johnson CR, Zhang B (1995) Solid phase synthesis of alkenes using the Horner–Wadsworth–Emmons reaction and monitoring by gel phase 31P NMR. Tetrahedron Lett 36(51):9253–9256
Paul S, Gupta M (2005) A simple and efficient method for selective single aldol condensation between arylaldehydes and acetone. Synth Commun 35:213–222
Pihko PM, Salo TM (2003) Excess sodium ions improve Z selectivity in Horner–Wadsworth–Emmons olefinations with the Ando phosphonate. Tetrahedron Lett 44:4361–4364
Rathke MW, Nowak M (1985) The Horner–Wadsworth–Emmons modification of the Wittig reaction using triethylamine and lithium or magnesium salts. J Org Chem 50:2624–2626
Reinhardt D, Ilgen F, Kralisch D, König B, Kreisel G (2008) Evaluating the greenness of alternative reaction media. Green Chem 10:1170–1181
Roelfs JCAA, Dillen AJ, Jong KP (2000) Base-catalyzed condensation of citral and acetone at low temperature using modified hydrotalcite catalysts. Catal Today 60:297–303
Sano S, Yokoyama K, Shiro M, Nagao Y (2002) A facile method for the stereoselective Horner–Wadsworth–Emmons reaction of aryl alkyl ketones. Chem Pharm Bull 50(5):706–709
Sano S, Takemoto Y, Nagao Y (2003) (E)-Selective Horner–Wadsworth–Emmons reaction of aryl alkyl ketones with bis(2,2,2-trifluoroethyl)phosphonoacetic acid. Tetrahedron Lett 44:8853–8855
Tichit D, Coq B, Cerneaux S, Durand R (2002) Condensation of aldehydes for environmentally friendly synthesis of 2-methyl-3-phenyl-propanal by heterogeneous catalysis. Catal Today 75:197–202
Tundo P, Anastas P, Black DS, Breen J, Collins T, Memoli S, Miyamoto J, Polyakoff M, Tumas W (2000) Synthetic pathways and processes in green chemistry: introductory overview. Pure Appl Chem 72:1207–1228
Wadsworth WS (2005) Synthetic applications of phosphoryl-stabilized anions. Org React 25(2):73–253
Wadsworth WS, Emmons WD (1961) The utility of phosphonate carbanions in olefin synthesis. J Am Chem Soc 83(7):1733–1738
Zumbansen K, Dçhring A, List B (2010) Morpholinium trifluoroacetate-catalyzed aldol condensation of acetone with both aromatic and aliphatic aldehydes. Adv Synth Catal 352:1135–1138
Acknowledgments
Financial support from the Research Council of Yazd University is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jafari, A.A., Ghadami, M. Efficient synthesis of α,β-unsaturated ketones with trans-selective Horner–Wadsworth–Emmons reaction in water. Environ Chem Lett 14, 223–228 (2016). https://doi.org/10.1007/s10311-016-0552-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10311-016-0552-8