
Abstract
Biological aerosol particles are recently gaining increased attention, but global estimates of their emissions are unclear. Traditional organic tracers for biological particles have not been able to capture the diversity of biological sources such as residential wood burning. Here, we have analyzed protein content in air particulate matter over a year in a suburban area near Shibuya, in order to identify contributions from biological sources. Aerosols were separated into five size fractions using a high-volume cascade impactor. Amino acid profiles were obtained from acid hydrolysates by high-performance liquid chromatography and were subsequently used in a receptor model for source apportionment. Results show that protein content ranged from 0.5 to 2 % throughout the year in all size fractions. We observed contributions from seasonally variable emission sources such as pollen and fallen leaves in coarse particles, larger than 7 µm, at elevated concentrations during spring and winter. We also found contributions from pollen and fallen leaves in the smallest size fraction, lower than 1.1 µm, after spring. This finding indicates that sources are broken up to smaller particles in the urban environment. Overall, our findings demonstrate that protein content can be used as marker for biological aerosol particles, and that protein amino acid composition can distinguish multiple sources.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Size of terrestrial primary biological aerosol particles is generally categorized as consisting mostly of coarse mode particles with a mass-based mode maximum at 2–3 µm. Even though primary biological aerosol particles smaller than 2 µm have long been recognized as sources of organic particulate matter (Matthias-Maser and Jaenicke 1993), they have enjoyed less attention than other anthropogenic or geologic sources. Recent studies on such small biological particles report significant contributions of fungal spores to urban atmospheric aerosol (Bauer et al. 2008) as well as the presence of bacteria and other microorganisms in cloud water samples (Bauer et al. 2003). Exposure to biological aerosol particles, especially of respirable size, is associated with a variety of health effects including infectious diseases, respiratory diseases, and cancer (Douwes et al. 2003). Primary biological aerosol particles are also relevant in cloud processes and may play an important role in global climate changes. They can more easily act as condensation or ice nuclei than most other aerosol particles, and large biological particles are suggested to act as so-called giant cloud condensation nuclei which facilitate rain formation by quickly growing to large sizes through coalescence with other droplets (Després et al. 2012).
Previous studies mostly concentrated on tracer compounds of specific source types such as allergens found in pollen and its fragments (Swoboda et al. 2004), or on dust deriving from pets (Holmquist and Vesterber 2002), households (Custovic et al. 1999), and roads (Miguel et al. 1999). Protein amino acids are found in most biological materials and can serve as a less specific marker which is able to capture a wide range of primary biological aerosol particles at once. Unlike cultivation methods applied for bacteria or fungi, where culturability is influenced by meteorological and experimental factors as well as by impaction stress in sampling devices (Després et al. 2012), protein amino acid compositions are relatively stable in aerosol samples. Their analysis is less time-consuming than microscopic classification methods (Matthias-Maser and Jaenicke 1993) but retains information on emission sources. Amino acid compositions, which have been measured in a few studies from atmospheric aerosols (Franze et al. 2003) or residential aerosols (Armstrong et al. 2001), are analyzed in a source–receptor model to identify contributing source types. Sampling periods covering a whole year provide new insight into seasonal variations in size distribution and composition of protein matter in the urban atmosphere.
Experimental
Sampling of aerosols
Ambient aerosol samples were collected on the rooftop of the three-story Komaba Communication Plaza building at the Komaba campus of the University of Tokyo, 1.5 km west of Shibuya, using a high-volume aerosol sampler equipped with an Andersen-type cascade impactor (AH-600F, Shibata Scientific Technology; equivalent to an Andersen Model 65-800) which separated aerosol samples into five size fractions of larger than 7, 7–3.3, 3.3–2, 2–1.1 µm, and smaller than 1.1 µm at a flow rate of 566 L min−1. Perforated Pallflex® Tissuquartz™ filters for the cascade impactor and backup filters (AHQ-630: ø = 30 cm; AHQ-640 8 × 10″) were purchased from Tokyo Dylec Corp. Filters were weighed in a humidity-controlled dust-free tent. Sampling times from 1 to 2 weeks covered a whole year ranging from February 2009 to January 2010 and yielded tens to hundreds of mg of particulate matter (PM) per filter of which an eighth part is used for analysis. Direct sampling from aerosol sources generally results in sufficiently high yields of fine PM within reasonably short sampling times, but was only successful for kitchen aerosols, representing a major source or organic aerosols in the urban environment (Rogge et al. 1991), and for pollen from Japanese cedar, the most common wind pollinating tree (Ministry of Agriculture, Forestry and Fisheries 2012) releasing the most important seasonal allergen in Japan. Sampling of fine particles from aerosols of other biological materials proved more difficult because they mainly consist of large particles, need to be transported, and ground down on widespread areas such as roads (Patra et al. 2008) to produce fine particles or only break down to fragments after aging or exposure to high humidity conditions. Simple grinding experiments of bulk samples did not produce enough fine particles for laboratory-scale re-suspension experiments with the aerosol sampler and were analyzed from powdered bulk samples. These samples included fallen leaves as well as skin and fingernail scrapings for the source group of keratins representing re-suspension of plant debris from roads and particles from skin and textile wear, respectively.
Kitchen aerosols were sampled during lunchtime from a kitchen exhaust vent of a pub in the vicinity of the campus, where chicken cutlets and fried chicken were continuously prepared. Aerosol was sampled through a duct (ø = 10 cm) from an unfiltered ventilation blower which discharged kitchen air at an average velocity of 3.0 m s−1 (ø = 30 cm). Pollen aerosols were directly sampled from some seven blooming Japanese cedar of the Kumodōri variety planted at the seedbed site at the University of Tokyo Chichibu Forest during March 2010. A cyclone separator was inserted upstream of the aerosol sampler in order to make sampling of aerosol samples of pollen possible, which would otherwise overload the cascade impactor with large particles. The cyclone separator with a similar geometry to the sharp cut cyclone (cutoff d p,50: 2.5 µm; flow rate: 16.7 L min−1; diameter: 3.495 cm) (Kenny et al. 2000) was scaled up to a diameter of 24 cm with a nominal cutoff diameter d p,50 of 7 µm at a flow rate of 566 L min−1, according to semiempirical equations by Lapple (1951).
Chemical analysis procedure
Protein content in aerosol and powdered samples were measured by analyzing fluorescent derivates of amino acids found after 24 h of vacuum hydrolysis at 110 °C in a heat block. Aliquots of filter or powder samples were subjected to hydrolysis with 6 ml of 6 M HCl following a procedure described elsewhere (Liu et al. 1995). Norleucine was added as internal standard before hydrolysis. After drying the filtered hydrolysate by rotary evaporation, its residue was dissolved in distilled water and promptly derivatized with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate solution from the AccQ-Tag™ reagent kit (Waters, Milford). Hydrolysis in 6 M HCl results in the conversion of the amide group in asparagine and glutamine to their respective acids (Perrot et al. 2002) and the destruction of tryptophan (Friedman and Finley 1971). The analysis therefore included only the remaining 17 canonical amino acids. This reduction in variables available for source apportionment may decrease the robustness of detected contributions for sources with otherwise similar amino acid compositions.
The high-performance liquid chromatography system from Shimadzu was operated at a temperature of 37 °C with an Inertsil® ODS-3 column (250 mm × 4.6 mm i.d.; 5 µm particles) and an RF-10AXL fluorescence detector (λ Ex = 250 nm, λ Em = 395 nm). Analysis of amino acids derivatized with 6-aminoquinolyl-N-hydroxysuccinimityl carbamate has been reported previously with fixed pH of 4.95–5.9 (Liu et al. 1995) or following a pH gradient (van Wandelen and Cohen 1997). However, large peaks from side reactions with H2O solvent and NH3 present in some samples could only be separated from proline and cysteine, respectively, by running a double analysis with the following two gradient programs:
Solvent A was a buffer solution of 180 mmol L−1 acetic acid/sodium acetate at pH 5.1 with 4 mmol L−1 trimethylamine additive. Solvents B and C were acetonitrile and distilled water, respectively, and total flow rate was 1 mL min−1. The gradient programs started with 100 %, v/v Solvent A and increased Solvent B percentages at two or three gradient slopes after 3 min. In Program 1, two slopes were applied for Solvent B at 0.8 % min−1 from 0 to 5 %, v/v and 1.6 % min−1 from 5 to 35 %, v/v. Program 2 used 0.8 % min−1 from 0 to 5 %, v/v, a slow gradient of 0.4 % min−1 from 5 to 10 % and 1.5 % min−1, v/v from 10 to 35 %. The column was flushed for 4 min after each analysis with 60 %, v/v acetonitrile (Solvent B)/distilled water (Solvent C) before reconditioning with Solvent A.
Receptor modeling
Contributions from primary biological aerosol sources to amino acid (protein) concentrations were estimated from ambient amino acid concentration data and source profiles using the chemical mass balance model by the US EPA (United States Environmental Protection Agency 2005), which finds wide use in aerosol source inventories, e.g., in Europe (Puxbaum 2011). Amino acid profiles have not been used as marker for primary biological aerosol particles in previous research. Analysis results of aerosol and bulk samples resembling primary biological aerosol emission sources were used as emission profiles. Kitchen exhaust and pollen from Japanese cedar trees were directly sampled with the aerosol sampler in order to observe possible differences in amino acid compositions between size fractions. For fallen leaves, keratins, silk fibroin, and fungal spores, amino acid profiles were compiled from analysis of powdered samples and data mainly found in the literature about nutritional value (see next section for details). Each of the source groups consisting of almost identical compositions was represented by an average amino acid profile with standard deviations as species uncertainties. Microbiological aerosol particle sources included only a reference for fungal spores of a single species and could not be extended within this study to take other sources such as bacteria into account.
Results and discussion
Amino acid concentrations in particulate matter
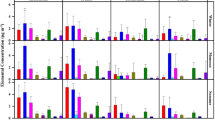
Amino acids measured from hydrolysate of urban ambient aerosol were detected in the range of less than one to several tens of ng m−3, as seasonal averages in Fig. 1. They are grouped by their side chains’ properties at pH 7 as hydrophilic with positive charge, negative charge, and uncharged as well as special and hydrophobic. Methionine, tyrosine, histidine, and tyrosine showed very low abundance and were often not detected or were not quantifiable in samples with low PM load (limit of quantification was around 1 ng m−3 amino acid for one-week samples). Asparagine, glutamine, and tryptophan could not be measured either due to conversion to acidic forms or for the latter due to decomposition during hydrolysis as mentioned in the previous section.

Seasonal averages of size-resolved amino acid concentrations in urban ambient aerosol. Amino acids of varying composition are found in PM throughout the year only with lower abundance in summer. *Asparagine, glutamine, and tryptophan could not be analyzed with the current method
Combined amino acid content is not limited to specific size fractions, but is found in all size fractions at nearly the same order of magnitude and makes up 0.5–2 % of PM (Fig. 2). This proportion showed no distinct seasonal variation though concentrations were somewhat lower in particles smaller than 1.1 µm during summer. Because of PM concentration was low in some samples and size fractions, only a selection has been analyzed. Spring and autumn show some similarity with high concentrations of lysine, valine, and proline in the larger size fractions. Overall protein fractions measured in Tokyo are lower than previous reports from Europe by Franze et al. (2003), but differences in amino acid compositions were observed not only for different episodes, but also for size fractions within a single sampling period. These compositions are analyzed after the next subsection on emission sources in source apportionment.

Combined amino acid content in ambient aerosol makes up about 0.5–2 % of PM. No trends could be observed in this proportion between particle-size fractions or observed PM concentrations
Aerosol source profiles
Amino acid concentrations in aerosol sampled from kitchen exhaust were highest with 6.1 µg m−3 amino acids in 267 µg m−3 for the smallest size fraction < 1 µm and showed similar concentrations of 2.0–2.8 µg m−3 in 59–122 µg m−3 PM found in larger size fractions. The amino acid pattern observed for the smallest particles in Fig. 3a is distinctively different from those in larger PM fractions with high content of glutamic acid and valine and may be influenced by Japanese seasoning. Amino acids make up 1.7–3.9 % of PM in each fraction. In pollen samples, large particles which make up 99.7 % of total PM (0.53 g m−3) at the time of emission were removed with the cyclone separator from particles smaller than 7 µm before analysis with the cascade impactor. PM could only be quantified on the first impactor stage collecting particles larger than 7 µm and on the backup filter (smaller than 1.1 µm), both at a concentration of only 5 mg in 8 m3 aerosol sample. Intermediate fractions (containing less than 2 mg per filter) were not further analyzed. Amino acid compositions in the two analyzed fractions shown in Fig. 3b were very similar with only minor differences and overall amino acid content made up 2.6 % of PM. Remarkable are the high concentrations of cysteine and valine. Results from these two fractions are used as source profiles for the two fractions larger than 3.3 µm and the remaining three fractions smaller than that, respectively.

Source profiles of a kitchen exhaust aerosol, b small-sized fractions of airborne pollen from cryptomeria japonica field samples, and of c fallen leaves, d keratins, e silk, and f fungal spores are used to explain ambient aerosol data. *Asparagine, glutamine, and tryptophan were not included in the receptor model
The remaining source profiles were pooled from multiple samples and literature data, where possible. Amino acid compositions of four dry fallen leaf samples from local maple and zelkova serrata (elm family) trees showed almost identical compositions and were combined as leaves in Fig. 3c. Amino acid content varied from 1 to 4 %, though. Further, tryptophan which is usually lost during hydrolysis has been detected in all leaf samples. Analysis results from skin scrapings and fingernail samples agreed with previous results for hair (Robbins 2012), wool (Cornfield and Robson 1955), horn, and feathers (Block and Bolling 1951) and were combined as keratins in Fig. 3d. Amino acid compositions in Fig. 3e, f show compositions obtained from the literature for silk fibroin (Shaw 1964) and fungal spores (Bent and Morton 1963), respectively.
Source apportionment
Source contributions estimated by the chemical mass balance model are shown for each size fraction from (a) with particles larger than 7 µm to (e) below 1.1 µm as colored bars in Fig. 4. Contributions from kitchen aerosols are present in all fractions but are only high in coarse particles larger than 7 µm (a) and for a few samples also in intermediate fractions (b) to (d) (1.1–7 µm). Most likely false assignments are seen in the sample starting on March 17, which should be dominated by eolian dust from Mongolian desert storms, as reported on 2 days for Tokyo (Japan Meteorological Agency 2009). Pollen is found in fraction (a) larger than 7 µm from February to March which represents the main pollen season in Japan with the Japanese cedar blooming. Leaves start occurring in fraction (a) larger than 7 µm around December, shift to fraction (e) smaller than 1.1 µm during February and are detected until around May. Keratins, silk, and spores did not show obvious seasonal trends and were detected in fraction (a) and (b) larger than 3.3 µm as well as in fraction (e) smaller than 1.1 µm. These results demonstrate that emission sources of primary biological aerosol particle can be traced by analyzing protein amino acid compositions down to the sub-micrometer region which previously required identification and analysis of a tracer substance for each source or time-consuming microscopic counting methods. In recent source apportionments of organic matter in PM2.5, e.g., for Paris (Crippa et al. 2013) or for the Los Angeles basin (Heo et al. 2013), primary biological aerosol sources remain unidentified or are insufficiently represented in factors such as cooking-related and wildfire-related aerosols. Including analysis of amino acids in such studies may allow several new types of primary biological aerosol particles to be distinguished, but also consumes a significant amount of sample.

Source apportionment results for aerosol size fractions a–e (>7, 7–3.3, 3.3–2, 2–1.1, and <1.1 µm) collected over varying sampling intervals from February 2009 to January 2010. Filled bars represent estimated contributions of each source group to total measured amino acid mass (empty bar) in each size fraction. Bars are left out for where sample analysis has been skipped
Conclusion
Protein content of primary biological aerosol particles has been utilized to detect their presence in five particle-size fractions separated by a high-volume cascade impactor. Protein amino acids are found in proportions of 0.5–2 % relative to PM almost uniformly distributed over all size fractions of urban aerosol down to particle sizes smaller than 1.1 µm. For the first time, seasonal variation in contributions from primary biological aerosol particle sources could be traced using their amino acid profiles without the need to use source-specific tracer substances, immunological or microscopic methods.
References
Armstrong DW, Kullman JP, Chen X, Rowe M (2001) Composition and chirality of amino acids in aerosol/dust from laboratory and residential enclosures. Chirality 13:153–158
Bauer H, Giebl H, Hitzenberger R, Kasper-Giebl A, Reishl G, Zibuschka F, Puxbaum H (2003) Airborne bacteria as cloud condensation nuclei. J Geophys Res 108(D21):4658
Bauer H, Schueller E, Weinke G, Berger A, Hitzenberger R, Marr IL, Puxbaum H (2008) Significant contributions of fungal spores to the organic carbon and to the aerosol mass balance of the urban atmospheric aerosol. Atmos Environ 42:5542–5549
Bent KJ, Morton AG (1963) Amino acid composition of fungi during development in submerged culture. Biochem J 92:260–269
Block RJ, Bolling D (1951) The amino acid composition of proteins and foods, 2nd edn. Charles C Thomas, Springfield
Cornfield MC, Robson A (1955) The amino acid composition of wool. Biochem J 59(1):62–68
Crippa M, DeCarlo PF, Slowik JG, Mohr C, Heringa MF, Chirico R, Poulain L, Freutel F, Sciare J, Cozic J et al (2013) Wintertime aerosol chemical composition and source apportionment of the organic fraction in the metropolitan area of Paris. Atmos Chem Phys 13:961–981
Custovic A, Woodcock H, Craven M, Hassall R, Hadley E, Simpson A (1999) Dust mit allergens are carried on not only large particles. Pediatr Allergy Immunol 10:258–260
Després VR, Huffman JA, Burrows SM, Hoose C, Safatov AS, Buryak G, Fröhlich-Novwoisky J, Elbert W, Andreae MO, Pöschl U et al (2012) Primary biological aerosol particles in the atmosphere: a review. Tellus Ser B. doi:10.3402/tellusb.v64i0.15598
Douwes J, Thorne P, Pearce N, Heederik D (2003) Bioaerosol health effects and exposure assessment: progress and prospects. Ann Occup Hyg 47(3):187–200
Franze T, Krauser K, Niessner R, Pöschl U (2003) Proteins and amino acids in air particulate matter. J Aerosol Sci 34(Supplement 2):S777–S778
Friedman M, Finley JW (1971) Methods of tryptophan analysis. J Agric Food Chem 19(4):626–631
Heo J, Dulger M, Olson MR, McGinnis Jerome SB, Sioutas C, Schauer JJ (2013) Source apportionments of PM2.5 organic carbon using molecular marker Positive Matrix Factorization and comparison of results from different receptor models. Atmos Environ 73:51–61
Holmquist L, Vesterber O (2002) Direct on air sampling filter quantification of cat allergen. J Biochem Biophys Methods 51:17–25
Japan Meteorological Agency (2009) Kosa (aeolian dust). Retrieved from http://www.data.kishou.go.jp/obs-env/kosahp/kosa_data_index.html
Kenny LC, Gussman R, Meyer M (2000) Development of a sharp-cut cyclone for ambient aerosol monitoring applications. Aerosol Sci Technol 32:338–358
Lapple CE (1951) Processes use many collector types. Chem Eng 58:144–151
Liu HJ, Chang BY, Yan HW, Yu FH, Liu XX (1995) Determination of amino acids in food and feed by derivatization with 6-aminoquinoyl-N-hydroxysuccinimidyl carbamate and reversed-phase liquid chromatographic separation. J AOAC Int 78(3):736–744
Matthias-Maser S, Jaenicke R (1993) Examination of atmospheric bioaerosol particles with radii > 0.2 µm. J Aerosol Sci 25(8):1605–1613
Miguel AG, Cass GR, Glovsky MM, Weiss J (1999) Allergens in paved road dust and airbornde particles. Environ Sci Technol 33:4159–4168
Misitry of Agriculture, Forestry and Fisheries (2012) Current status of forest resources. Retrieved from http://www.rinya.maff.go.jp/j/keikaku/genkyou/index1.html
Patra A, Colvile R, Arnold S, Bowen E, Shallcross D, Martin D, Price C, Tate J, ApSimon H, Robins A (2008) On street observations of particulate matter movement and dispersion due to traffic on an urban road. Atmos Environ 42:3911–3926
Perrot L, Charpentier M, Charpentier C, Feuillat M, Chassagne D (2002) Yeast adapted to wine: nitrogen compounds released during induced autolysis in a model wine. J Ind Microbiol Biotechnol 29:134–139
Puxbaum H (2011) Aerosol source-receptor studies. In: Puxbaum H, Winiwarter W (eds) Advances of atmospheric aerosol research in Austria. Austrian Academy of Sciences, Vienna, pp 89–107
Robbins CR (2012) Chemical and physical behavior of human hair. Springer, Berlin
Rogge WF, Hildemann LM, Mazurek MA, Cass GR (1991) Sources of fine organic aerosol. 1. Charbroilers and meat cooking operations. Environ Sci Technol 25:1112–1125
Shaw J (1964) Fractionation of fibroin of Bombyx mori with alkali. Biochem J 93:54–61
Swoboda I, Grote M, Verdino P, Keller W, Singh MB, De Weerd N, Sperr WR, Valent P, Balic N, Reichelt R et al (2004) Molecular characterization of polygalacturonases as grass pollen-specific marker allergens: expulsion from pollen via submicronic respirable particles. J Immunol 172(10):6490–6500
United States Environmental Protection Agency (2005) Chemical mass balance (CMB) model. Retrieved from http://www.epa.gov/scram001/receptor_cmb.htm
van Wandelen C, Cohen SA (1997) Using quaternary high-performance liquid chromatography elution systems for separating 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate-derivatized amino acid mixtures. J Chromatogr A 763:11–22
Acknowledgments
Thanks go to the Komaba Communication Plaza for providing an aerosol sampling location for over a year, The University of Tokyo Chichibu Forest for access to blooming Japanese cedars as well as Itamae-Ryōri Ebisuya for providing unfiltered kitchen aerosol.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Abe, R.Y., Akutsu, Y. & Kagemoto, H. Protein amino acids as markers for biological sources in urban aerosols. Environ Chem Lett 14, 155–161 (2016). https://doi.org/10.1007/s10311-015-0536-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10311-015-0536-0