
Abstract
Clostridium thermocellum is a potentially useful organism for the production of lignocellulosic biofuels because of its ability to directly deconstruct cellulose and convert it into ethanol. Previously engineered C. thermocellum strains have achieved higher yields and titers of ethanol. These strains often initially grow more poorly than the wild type. Adaptive laboratory evolution and medium supplementation have been used to improve growth, but the mechanism(s) by which growth improves remain(s) unclear. Here, we studied (1) wild-type C. thermocellum, (2) the slow-growing and high-ethanol-yielding mutant AG553, and (3) the faster-growing evolved mutant AG601, each grown with and without added formate. We used a combination of transcriptomics and proteomics to understand the physiological impact of the metabolic engineering, evolution, and medium supplementation. Medium supplementation with formate improved growth in both AG553 and AG601. Expression of C1 metabolism genes varied with formate addition, supporting the hypothesis that the primary benefit of added formate is the supply of C1 units for biosynthesis. Expression of stress response genes such as those involved in the sporulation cascade was dramatically over-represented in AG553, even after the addition of formate, suggesting that the source of the stress may be other issues such as redox imbalances. The sporulation response is absent in evolved strain AG601, suggesting that sporulation limits the growth of engineered strain AG553. A better understanding of the stress response and mechanisms of improved growth hold promise for informing rational improvement of C. thermocellum for lignocellulosic biofuel production.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alternative forms of energy ranging from solar and wind power to nuclear fusion are being investigated as potential replacements to end human dependence on fossil fuels. Even with anticipated advances in electrical and battery technologies, liquid fuels will likely continue to be required for long-range shipping and transport. For liquid fuel production, one of the most promising options and potentially cost-effective technologies [21] involves is the conversion of lignocellulosic biomass into fuels and chemicals by microorganisms in a process called Consolidated bioprocessing (CBP). With CBP, microbial systems directly deconstruct and convert lignocellulosic plant material into value-added compounds in a single pot without the addition of cellulolytic enzymes [7].
Clostridium thermocellum is one of the well-equipped organisms for implementing a CBP strategy for bioconversion. It is a Gram-positive, thermophilic Firmicute that utilizes a multi-enzyme complex called the cellulosome to naturally depolymerize cellulose and hemicellulose [2]. Though C. thermocellum naturally deconstructs lignocellulosic biomass and produces the biofuel ethanol, it also generates a number of unwanted fermentation products including lactate, formate, acetate, hydrogen, and amino acids [9, 14], all of which limit the microbe’s industrial utility as carbon flux is diverted away from the desired product. Metabolic engineering is, thus, crucial to controlling carbon and electron flux to only the product of interest, and substantial effort has gone into genetically modifying C. thermocellum for enhanced ethanol production [1, 4, 6, 8, 22, 23, 28].
Unfortunately, engineering C. thermocellum central metabolism has, in many cases, resulted in poor growth [6, 22, 23], which is another barrier to industrial deployment. Deletion of the hydrogenase maturation protein (hydG) resulted in poor growth, but medium supplementation with acetate improved its growth rate [5]. Additionally, pyruvate:formate lyase (pfl) was deleted in C. thermocellum, which tripled the doubling time of the strain in comparison to the wild type [23]. When a C. thermocellum ∆pfl strain was grown in a medium supplemented with formate, the doubling time improved from approximately 6 to 3.4 h. This corroborated a previous study in Staphylococcus aureus where supplementing the growth medium with formate after the deletion of pfl corrected much of the growth defect, suggesting that the elimination of formate production inhibited growth due to the disruption of C1 metabolism [15]. Formate can be ligated to tetrahydrofolate (THF) to produce formyl-THF, which in turn provides the formyl-, methylene- and methyl groups needed for the biosynthesis of serine, purine, formyl-methionine, methionine and S-adenosyl-methionine [26]. A similar need for C1-units for biosynthesis was proposed as an explanation for the growth defect in C. thermocellum ∆pfl [23], but this hypothesis has not yet been tested.
A slow growth phenotype was observed in C. thermocellum strain AG553, in which the hydG, pfl, lactate dehydrogenase (ldh) and phosphotransacetylase-acetate kinase (pta-ack) genes are deleted, resulting in a strain where metabolic pathways to acetate, lactate, formate, and most H2 were removed. Strain AG553 exhibited substantially higher ethanol yields than the wild-type strain, but a growth rate of 0.13/h in minimal medium compared with a growth rate of 0.25/h for the wild-type strain [27]. For an industrial setting, both ethanol yield and productivity are crucial, making the growth defect problematic.
To overcome slow growth, adaptive laboratory evolution was applied to AG553. The strain was evolved for ~ 1500 generations with 5 g/L cellobiose as the growth substrate, resulting in the isolated strain AG601 [27]. During the evolution from strain AG553 to AG601, mutations were acquired that were hypothesized to be responsible for the improved growth phenotype by allowing continued sugar metabolism under stressful conditions that might otherwise have caused sporulation [27]. Therefore, one of the mutations most likely to be involved in improved growth and higher ethanol titer is related to sporulation. Clostridium thermocellum is atypical in that it has two copies of the canonical master regulator of sporulation, Spo0A (Clo1313_0637 and Clo1313_1409). Clo1313_1409 was found to be essential for sporulation, and deleting it eliminated sporulation [20]. Strain AG601, on the other hand, has a mutation in Clo1313_0637, as does a mutant strain of C. thermocellum strain ATCC27405 that was evolved to be tolerant to poplar hydrolysate [16], where the ortholog Cthe_3087 (99% protein sequence identity) acquired a mutation. Another potential gene involved in the growth difference encodes the transcription termination protein Rho, and a similar mutation in E. coli resulted in transcriptional read through, altered gene expression, and increased tolerance to ethanol [12].
Robust growth and fermentation are an essential part of any bioconversion process, and a deeper understanding of the physiology of both parent and mutant strains is critical to rationally improve the robustness of these organisms. Because ΔhydG and Δpfl are part of the genetic background in both the engineered strain AG553 and the evolved strain AG601, it stood to reason that medium supplementation could benefit the growth of these strains in the same way it did for the individual mutants. Likewise, elucidating the systematic response imparted by supplementation, via transcriptomic and proteomic measurements, could reveal new genetic targets to improve growth.
Materials and methods
Growth media
Clostridium thermocellum was grown in defined Media for Thermophilic Clostridia (MTC) [13] modified to reduce the urea concentration and filter-sterilized to avoid autoclaving. This modified defined media are referred to as MTC5 [22]. Where indicated, MTC5 was supplemented with 5 mM sodium acetate or 2 mM sodium formate.
Cultures for proteomics and RNA sequencing
Cultures of C. thermocellum were revived from − 80 °C freezer stocks into 5 mL MTC5 at 51 °C in a Coy Anaerobic Chamber (Coy Laboratory Products, Grass Lake, MI, USA) and grown to mid-exponential phase (OD ~ 0.4) in triplicate. 2 mL of the growing culture was added to 100 mL MTC5 in a 298-mL serum bottle sealed with a butyl rubber stopper (Chemglass Life Sciences, Vineland, NJ). Cultures were grown to exponential phase (OD ~ 0.27) at which point they were rapidly transferred to two 50-mL conical tubes and centrifuged at 8000 rpm for 5 min at 4 °C. The supernatant was removed, and the cultures were immediately flash frozen in liquid N2. Frozen cell pellets were held at − 80 °C until protein or RNA extraction was performed.
RNA isolation and ribosomal RNA removal
RNA was isolated and depleted of rRNA essentially as described previously [29]. Briefly, cell pellets were resuspended in 1 mL of TRIzol (Invitrogen, Carlsbad, CA, USA) and lysed by bead beating with 0.8 g of 0.1-mm glass beads (BioSpec Products, Bartlesville, OK, USA) for 3 × 20 s each at 6500 rpm in a Precellys 24 high-throughput tissue homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France). The RNA from each cell lysate was purified using Qiagen RNeasy Mini kit, which incorporated a DNaseI treatment on the purification column, in accordance with the manufacturer’s instructions. Purified RNA was quantified and assessed for quality using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and an Agilent Bioanalyzer (Agilent, CA, USA). High-quality total RNA (RIN > 8) was depleted of rRNA using Ribo-Zero rRNA Removal Kit for bacteria (Epicentre, San Diego, USA) following the manufacturer’s protocol. The depleted sample was purified on a RNA Clean & Concentrator-5 (Zymo Research, Irvine, CA, USA) following the manufacturer’s protocol.
Library preparation and sequencing
Depleted RNA was used for RNA-Seq library preparation with the Epicentre ScriptSeq v2 RNA-Seq Library Preparation Kit (Epicentre, San Diego, CA, USA) following the manufacturer’s protocol (EPILIT329 Rev.C). Agencount AMPure beads (Beckman Coulter, Indianapolis, USA) were used to purify the cDNA, and unique indexes were added during 13 cycles of library amplification. Final RNA-Seq libraries were purified with Agencount AMPure beads (Beckman Coulter, Indianapolis) and quantified with a Qubit fluorometer (Life Technologies, Carlsbad, CA, USA). The library quality was assessed on a Bioanalyzer DNA 7500 DNA Chip (Agilent, Santa Clara, CA, USA), and samples were pooled and diluted. Sequencing was completed using a SR50 sequencing protocol on an Illumina HiSeq 2500 platform (HudsonAlpha Genomic Services Laboratory; Huntsville, AL, USA).
RNA-Seq analysis
The analysis of the raw RNA-Seq data was performed as previously described [19]. Raw reads were mapped to the reference genome [10] [NCBI GenBank accession: CP002416] using CLC Genomics Workbench version 8.0 (CLC bio, Aarhus, Denmark) and the counts of uniquely mapped reads were analyzed for differential gene expression by DESeq 2 [17]. Filtering was applied to identify those genes with an FDR < 0.05 and fold changes of equal to or greater than a log2 of ± 1 for differential gene expression (Supplemental File 1). Raw RNA-Seq data have been deposited in NCBI Sequence Read Archive under accession SRP070784 and gene expression data under NCBI GEO accession GSE78287.
Proteomic sample preparation
Four washed cell pellets per condition (~ 50 mg each) of mid-log cultures were resuspended in lysis buffer (4% SDS, 100 mM Tris–HCl pH 8.0) and lysed via sonic disruption (Branson Sonifier). Crude lysates were then quantified by BCA, adjusted to a final concentration of 20 mM dithiothreitol (DTT), incubated at 100 °C for 10 min, and precleared via centrifugation at 21,000×g. Two milligrams of protein extract was precipitated with 20% trichloroacetic acid on ice, pelleted, washed with cold acetone, and air dried. Protein pellets were re-solubilized in 8-M urea, 5-mM DTT, 100-mM Tris–HCl, pH 8.0, treated with 15-mM iodoacetamide, and digested with trypsin as previously described [11]. Tryptic peptides were then salted (200 mM NaCl), acidified (0.1% formic acid), and passed through a 10-kDa MWCO size exclusion spin column to collect peptides of optimum size.
Tryptic peptides were quantified by BCA and 25 ug loaded via pressure cell onto a biphasic MudPIT column for online two-dimensional HPLC separation (strong-cation exchange and reversed phase) with concurrent nanospray MS/MS analysis using a hybrid LTQ-Orbitrap XL mass spectrometer (Thermo Scientific) operating in data-dependent acquisition (one full scan at 15-k resolution followed by 10 MS/MS scans in the LTQ, all one μscan; charge state screening (reject z = 0) with monoisotopic precursor selection; 2.2 m/z isolation window; dynamic exclusion: window = 20 ppm, duration = 30 s). Eleven salt cuts of ammonium acetate (25, 30, 35, 40, 45, 50, 65, 80, 100, 175, and 500 mM) were performed per sample run, each followed by a 100-min organic gradient to separate peptides.
Resulting MS/MS spectra were matched to computationally predicted tryptic peptides gleaned from the supplied proteome FASTA database [C. thermocellum 1313 protein sequences appended with common contaminants and reversed/decoy sequences to assess false-discovery rates (FDR)] using MyriMatch v. 2.1 [24]. Peptide spectrum matches (PSMs) were filtered by IDPicker v.3 [18] (2 peptide minimum per protein; peptide-level FDR < 1%) and assigned matched-ion intensities (MIT) based on observed peptide fragment peaks. PSM MITs were summed on a per-peptide basis, and only those uniquely and specifically matching a particular protein were moved onto subsequent analysis. Resulting peptide intensities were log2-transformed with the resulting log-normal distributions normalized across replicates (LOESS), and standardized across all samples by median absolute deviation and median centering using InfernoRDN [25]. Protein abundances were derived using the RRollup method as previously described [31]. Blank values were imputed with a normal distribution of low end values (downshift = 1.8, width = 3) using Perseus software to simulate limit-of-detection. Sample-to-sample variation was assessed by PCA and Pearson’s correlation analysis (Supplemental File 2). Protein abundances were then compared across strains and culture conditions to identify proteins with differential expression via one-way ANOVA via JMP Genomics software (Supplemental File 3).
Results
Formate addition increases growth rate of mutant strains
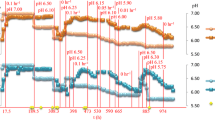
Medium supplementation with 5-mM acetate improved growth for the C. thermocellum hydrogenase maturase mutant strain ΔhydG, and the addition of 2 mM formate improved the growth of the C. thermocellum Δpfl. Because both hydG and pfl are deleted in strains AG553 and AG601, we hypothesized that medium supplementation with acetate and formate could improve growth of these strains as well. Therefore, we tested growth of wild type C. thermocellum, AG553, and AG601 when supplemented with acetate (Fig. 1a) or formate (Fig. 1b) at the concentrations previously demonstrated to elicit a physiological response. Little to no improvement in growth was seen upon addition of acetate for any of the strains. Formate supplementation, however, dramatically increased the growth rate of both AG553 and AG601, while no difference was observed in the wild type. To determine if the addition of formate altered flux through fermentation pathways, we quantified the fermentation products at the end of growth with and without added formate (Table 1). The addition of formate did not substantially change fermentation profiles, suggesting that the impact of formate addition is not at the level of fermentative pathway balancing.

Growth profiles for wild type C. thermocellum, AG553 and AG601 with and without the addition of a 5 mM acetate or b 2 mM formate. Growth curves represent the average of three cultures sampled every 15 min. A similar figure that includes error bars can be found in Supplemental Fig. 1. Black, Wild Type; Red, AG553; Blue, AG601. For all three strains, solid line, no supplementation; dotted line, with supplementation
Changes in gene expression after gene deletions
To gain insight into the deficiencies of these engineered strains and to gain a deeper understanding of the physiological benefits of formate supplementation, proteomics and RNA sequencing were performed with and without formate supplementation. Genes with a statistically significant (p < 0.05) change in expression of at least twofold were deemed differentially expressed (for complete dataset, see Supplemental File 1).
Comparison of un-evolved mutant strain AG553 to the wild-type strain in non-supplemented growth medium can help to elucidate system-wide changes brought upon by the deletion of the four central metabolism genes. Unsurprisingly, the deletion of these genes had a large effect on gene expression; under these conditions, 1184 genes were differentially expressed relative to wild type. Of these, 625 showed increased expression, while 559 decreased. Many of these differentially expressed genes were involved in tolerance to adverse conditions, with sporulation and stress-response genes being some of the most highly upregulated. In particular, Spo0A homolog Clo1313_1409 showed 37-fold higher expression in AG553 relative to the wild type. Among the downregulated genes, Clo1313_0108–Clo1313_0115, a cluster of genes involved in sulfur metabolism, show some of the greatest differences (6.5–13-fold difference). The serine hydroxymethyltransferase (SHMT; Clo1313_1155), which could be used either for serine biosynthesis or as a source of C1 units (Fig. 2), had 12.5-fold lower expression in AG553 relative to the wild type, which could impact C1 metabolism in this strain.

Statistically significant changes (p value < 0.05) in C1 metabolism observed in strain AG553 with and without added formate. Clo1313_#### gene numbers are named in parentheses and numbers associated indicate the log2 fold change seen in the RNA sequencing data. Positive numbers represent increased expression when formate is added. PRPP phosphoribosyl pyrophosphate, Atase amidophosphoribosyltransferase, GAR glycineamide ribonucleotide, FGAR phosphoribosyl-N-formylglycineamide, AIR aminoimidazole ribotide, AIR aminoimidazole ribotide, CAIR N5-carboxy-AIR, THF tetrahydrofolate, SHMT serine hydroxymethyltransferase, SAM S-adenosyl-methionine
Changes in gene expression after formate addition
In wild-type C. thermocellum, only two genes were differentially expressed upon addition of formate to the growth medium. Clo1313_0111 and Clo1313_0113 were both moderately downregulated (− 1.1 and − 1.2 log2 fold change, respectively) with the addition of formate. Both of these genes are predicted to be involved in siroheme biosynthesis.
To identify the effects of formate addition on the unevolved AG553, the RNA sequencing profiles of AG553 with and without formate supplementation were compared. After formate addition, 198 genes increased in expression and 153 decreased, including several genes involved in C1 metabolism. Five genes involved in converting phosphoribosyl-pyrophosphate (PRPP) into aminoimidazole ribotide for purine and thiamine biosynthesis (Clo1313_1009–Clo1313_1013) were all expressed more highly in the absence of formate by 2.0- to 2.8-fold (Fig. 2). The serine hydroxymethyltransferase expression level increases in AG553 in the presence of added formate, by 2.8-fold. A series of five genes (Clo1313_0109–Clo1313_0112) involved in siroheme biosynthesis, including the SAM-dependent methyltransferase cobA, were all significantly upregulated with the addition of formate in AG553. Both transcriptional and translational processes showed upregulation with the addition of formate to the AG553 strain, consistent with the faster growth rate. Comparing AG553 + formate with wild type revealed that expression of genes involved in sporulation and stress were still very prevalent in AG553, even after growth rate was improved by the addition of formate.
Changes in gene expression after adaptive laboratory evolution
To help identify the mechanisms by which growth improved after evolution of AG553, the evolved strain AG601 was compared with AG553 with no formate added. Strain AG601 showed 905 differentially expressed genes compared with AG553, of which 374 were more highly expressed after evolution. Most strikingly, the sporulation response is no longer prominent in AG601, with many sporulation-related genes dramatically (> 100-fold) more highly expressed prior to strain evolution (i.e., in strain AG553). Expression of the Spo0A homolog Clo1313_0637, which is mutated in AG601, increased 3.4-fold after evolution, while the other Spo0A homolog Clo1313_1409 decreased 28-fold.
In strain AG601 the impact of formate addition was far less dramatic than in strain AG553, with 234 genes differentially expressed upon addition of formate, of which 117 were more highly expressed with formate. Similar to strain AG553, C1-metabolism genes (Clo1313_1008–1013) were more highly expressed in the absence of formate. Unlike AG553, the translation machinery of AG601 was less differentially expressed in response to formate addition, consistent with the improved growth after evolution.
Proteome changes support transcriptome changes
Protein abundance changes were also measured in the same samples used for transcriptomics (for complete dataset, see Supplemental File 3). The overall trends in the proteomics data support those observed in the RNA sequencing data (Supplemental Fig. 2). The cluster of C1-metabolism genes Clo1313_1008–Clo1313_1013 all showed substantially higher abundances in both AG553 and AG601 in the absence of added formate relative to the wild type or the cultures with added formate. Similarly, the changes in mRNA abundance of genes associated with sporulation were also consistent in the proteomics dataset. For example, Spo0A homolog Clo1313_1409 was between 158- and 416-times more abundant in AG553 than in the wild-type strain, with or without formate addition, respectively, and Clo1313_1193 (stage IV sporulation protein) was on average 125 times more abundant in AG553 (relatively no difference observed attributed to formate addition). These proteins showed no statistically significant change in abundance when comparing the wild-type strain to AG601, also consistent with the transcriptomics data.
Discussion
Strain performance is critical for industrial biofuel production. Metrics to evaluate performance include not only the yield and titer of ethanol, but also the rate at which it is produced as well as the robustness of the strain. Though strains AG553 and AG601 are amongst the highest ethanol-yielding strains of C. thermocellum engineered to date, they exhibit a severely reduced growth rate compared to the wild type. Because both supplementation with formate and adaptive laboratory evolution helped to alleviate the growth defect in strain AG553, we also wanted to measure the system-wide impact of these approaches. The biggest impacts from formate addition were to C1 metabolism and the translation machinery. Strain evolution from AG553 to AG601, on the other hand, primarily prevented an aberrant sporulation response, presumably preventing metabolic shutdown under stressful conditions.
Formate addition greatly improved the growth rate of both strains of C. thermocellum containing the Δpfl mutation, but exogenous formate did not alter the fermentation end-product profiles in any of the strains studied. The lack of formate synthesis by the cell (i.e., ∆pfl) appears to generate a formate starvation response, with SHMT and the shared pathway to purine and thiamine metabolism (Clo1313_1008-1013) being upregulated, and SAM dependent proteins being downregulated. Previous isotopic labeling studies demonstrated that serine can be labeled with 13C-formate [30], suggesting that SHMT plays a role in C1 metabolism. Together, this provides direct support to the idea that C. thermocellum pfl mutants are deficient in C1 metabolism, and that overcoming this deficiency will improve growth and productivity, confirming our previous hypothesis [23]. Therefore, providing alternate genetic routes to enhance C1 metabolism is a promising route for future engineering of robustness in these strains.
In strain AG553, expression of ribosomal proteins and other translation-related genes did return to wild-type levels after the addition of formate, suggesting that these genes respond to growth rate, as has been observed in other organisms [3]. However, many of the stress response genes continued to be upregulated in the un-evolved strain. The fact that this stress response persists, even when biosynthetic needs are met, indicates that the deficiency in C1 metabolism is neither the only nor likely the primary source of stress in the cell. Therefore, the continued presence of a stress response is more likely to be caused by redox imbalances created by the deletion of multiple core fermentative pathways rather than the lack of metabolic building blocks.
The improved growth of strain AG601 is likely due to the lack of a sporulation response and corresponding shutdown of metabolism under stressful conditions. While it is possible that the previously identified mutation in the transcriptional terminator rho allows read through of unknown genes that dampen the sporulation response, a more likely explanation is that the known point mutation in the Spo0A homolog Clo1313_0637 causes the phenotype. The other Spo0A homolog, Clo1313_1409, is known to be essential for sporulation [20], and it is not detected in either wild type or AG601 in the proteomics dataset. However, this protein is very highly abundant in AG553 both with and without formate supplementation, consistent with the 37- and 28-fold higher transcript levels in AG553 relative to wild type and AG601, respectively. This lack of detectable Clo1313_1409 protein in the presence of the mutated Clo1313_0637 also suggests that the two Spo0A homologs work in tandem to control the sporulation response, where the mutated Clo1313_0637 is either inactive or constitutively active, shutting down Clo1313_1409. Future work will be needed to fully elucidate these interactions.
Conclusion
The high ethanol yields achieved in strains AG553 and AG601 were an important step toward creating an industrial CBP organism, but the slow growth phenotype is a severe limitation. Here, we utilized transcriptomics and proteomics to gain insight into the causes of the poor growth. By better understanding the nature of the stress response, the need for C1 donors for biosynthesis, and how strain evolution impacted each of these, this work will help to inform new strategies for engineering C. thermocellum for lignocellulosic biofuel production.
References
Argyros DA, Tripathi SA, Barrett TF, Rogers SR, Feinberg LF, Olson DG, Foden JM, Miller BB, Lynd LR, Hogsett DA, Caiazza NC (2011) High ethanol titers from cellulose using metabolically engineered thermophilic, anaerobic microbes. Appl Environ Microb 77:8288–8294. https://doi.org/10.1128/aem.00646-11
Bayer EA, Morag E, Lamed R (1994) The cellulosome—a treasure-trove for biotechnology. Trends Biotechnol 12:379–386. https://doi.org/10.1016/0167-7799(94)90039-6
Binder BJ, Liu YC (1998) Growth rate regulation of rRNA content of a marine Synechococcus (Cyanobacterium) strain. Appl Environ Microbiol 64:3346–3351
Biswas R, Prabhu S, Lynd LR, Guss AM (2014) Increase in ethanol yield via elimination of lactate production in an ethanol-tolerant mutant of Clostridium thermocellum. PLoS One 9:e86389. https://doi.org/10.1371/journal.pone.0086389
Biswas R, Wilson CM, Giannone RJ, Klingeman DM, Rydzak T, Shah MB, Hettich RL, Brown SD, Guss AM (2017) Improved growth rate in Clostridium thermocellum hydrogenase mutant via perturbed sulfur metabolism. Biotechnol Biofuels. https://doi.org/10.1186/s13068-016-0684-x
Biswas R, Zheng T, Olson DG, Lynd LR, Guss AM (2015) Elimination of hydrogenase active site assembly blocks H2 production and increases ethanol yield in Clostridium thermocellum. Biotechnol Biofuels 8:20. https://doi.org/10.1186/s13068-015-0204-4
Demain AL (2009) Biosolutions to the energy problem. J Ind Microbiol Biotechnol 36:319–332. https://doi.org/10.1007/s10295-008-0521-8
Deng Y, Olson DG, Zhou J, Herring CD, Joe Shaw A, Lynd LR (2013) Redirecting carbon flux through exogenous pyruvate kinase to achieve high ethanol yields in Clostridium thermocellum. Metab Eng 15:151–158. https://doi.org/10.1016/j.ymben.2012.11.006
Ellis LD, Holwerda EK, Hogsett D, Rogers S, Shao X, Tschaplinski T, Thorne P, Lynd LR (2012) Closing the carbon balance for fermentation by Clostridium thermocellum (ATCC 27405). Bioresour Technol 103:293–299. https://doi.org/10.1016/j.biortech.2011.09.128
Feinberg L, Foden J, Barrett T, Davenport KW, Bruce D, Detter C, Tapia R, Han C, Lapidus A, Lucas S, Cheng JF, Pitluck S, Woyke T, Ivanova N, Mikhailova N, Land M, Hauser L, Argyros DA, Goodwin L, Hogsett D, Caiazza N (2011) Complete genome sequence of the cellulolytic thermophile Clostridium thermocellum DSM1313. J Bacteriol 193:2906–2907. https://doi.org/10.1128/JB.00322-11
Giannone RJ, Wurch LL, Heimerl T, Martin S, Yang Z, Huber H, Rachel R, Hettich RL, Podar M (2015) Life on the edge: functional genomic response of Ignicoccus hospitalis to the presence of Nanoarchaeum equitans. ISME J 9:101–114. https://doi.org/10.1038/ismej.2014.112
Haft RJ, Keating DH, Schwaegler T, Schwalbach MS, Vinokur J, Tremaine M, Peters JM, Kotlajich MV, Pohlmann EL, Ong IM, Grass JA, Kiley PJ, Landick R (2014) Correcting direct effects of ethanol on translation and transcription machinery confers ethanol tolerance in bacteria. Proc Natl Acad Sci USA 111:E2576–2585. https://doi.org/10.1073/pnas.1401853111
Hogsett D (1995) Cellulose hydrolysis and fermentation by Clostridium thermocellum for the production of ethanol. Dartmouth College, Hanover
Holwerda EK, Thorne PG, Olson DG, Amador-Noguez D, Engle NL, Tschaplinski TJ, van Dijken JP, Lynd LR (2014) The exometabolome of Clostridium thermocellum reveals overflow metabolism at high cellulose loading. Biotechnol Biofuels 7:155. https://doi.org/10.1186/s13068-014-0155-1
Leibig M, Liebeke M, Mader D, Lalk M, Peschel A, Gotz F (2011) Pyruvate formate lyase acts as a formate supplier for metabolic processes during anaerobiosis in Staphylococcus aureus. J Bacteriol 193:952–962. https://doi.org/10.1128/JB.01161-10
Linville JL, Rodriguez M, Brown SD, Mielenz JR, Cox CD (2014) Transcriptomic analysis of Clostridium thermocellum Populus hydrolysate-tolerant mutant strain shows increased cellular efficiency in response to Populus hydrolysate compared to the wild type strain. BMC Microbiol. https://doi.org/10.1186/S12866-014-0215-5 (Artn 215)
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. https://doi.org/10.1186/s13059-014-0550-8 (ARTN 550)
Ma ZQ, Dasari S, Chambers MC, Litton MD, Sobecki SM, Zimmerman LJ, Halvey PJ, Schilling B, Drake PM, Gibson BW, Tabb DL (2009) IDPicker 2.0: improved protein assembly with high discrimination peptide identification filtering. J Proteome Res 8:3872–3881. https://doi.org/10.1021/Pr900360j
Manga P, Klingeman DM, Lu TY, Mehlhorn TL, Pelletier DA, Hauser LJ, Wilson CM, Brown SD (2016) Replicates, read numbers, and other important experimental design considerations for microbial RNA-seq identified using Bacillus thuringiensis Datasets. Front Microbiol 7:794. https://doi.org/10.3389/fmicb.2016.00794
Mearls EB, Lynd LR (2014) The identification of four histidine kinases that influence sporulation in Clostridium thermocellum. Anaerobe 28:109–119. https://doi.org/10.1016/j.anaerobe.2014.06.004
Olson DG, McBride JE, Shaw AJ, Lynd LR (2012) Recent progress in consolidated bioprocessing. Curr Opin Biotechnol 23:396–405. https://doi.org/10.1016/j.copbio.2011.11.026
Papanek B, Biswas R, Rydzak T, Guss AM (2015) Elimination of metabolic pathways to all traditional fermentation products increases ethanol yields in Clostridium thermocellum. Metab Eng 32:49–54. https://doi.org/10.1016/j.ymben.2015.09.002
Rydzak T, Lynd L, Guss A (2015) Elimination of formate production in Clostridium thermocellum. J Ind Microbiol Biotechnol 42:1263–1272. https://doi.org/10.1007/s10295-015-1644-3
Tabb DL, Fernando CG, Chambers MC (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res 6:654–661. https://doi.org/10.1021/Pr0604054
Taverner T, Karpievitch YV, Polpitiya AD, Brown JN, Dabney AR, Anderson GA, Smith RD (2012) DanteR: an extensible R-based tool for quantitative analysis of -omics data. Bioinformatics 28:2404–2406. https://doi.org/10.1093/bioinformatics/bts449
Thauer RK, Kirchniawy FH, Jungermann KA (1972) Properties and function of the pyruvate-formate-lyase reaction in clostridiae. Eur J Biochem 27:282–290
Tian L, Papanek B, Olson DG, Rydzak T, Holwerda EK, Zheng T, Zhou J, Maloney M, Jiang N, Giannone RJ, Hettich RL, Guss AM, Lynd LR (2016) Simultaneous achievement of high ethanol yield and titer in Clostridium thermocellum. Biotechnol Biofuels 9:116. https://doi.org/10.1186/s13068-016-0528-8
Tripathi SA, Olson DG, Argyros DA, Miller BB, Barrett TF, Murphy DM, McCool JD, Warner AK, Rajgarhia VB, Lynd LR, Hogsett DA, Caiazza NC (2010) Development of pyrF-based genetic system for targeted gene deletion in Clostridium thermocellum and creation of a pta mutant. Appl Environ Microb 76:6591–6599. https://doi.org/10.1128/AEM.01484-10
Wilson CM, Yang S, Rodriguez M Jr, Ma Q, Johnson CM, Dice L, Xu Y, Brown SD (2013) Clostridium thermocellum transcriptomic profiles after exposure to furfural or heat stress. Biotechnol Biofuels 6:131. https://doi.org/10.1186/1754-6834-6-131
Xiong W, Lin PP, Magnusson L, Warner L, Liao JC, Maness PC, Chou KJ (2016) CO2-fixing one-carbon metabolism in a cellulose-degrading bacterium Clostridium thermocellum. Proc Natl Acad Sci USA 113:13180–13185. https://doi.org/10.1073/pnas.1605482113
Xu Q, Resch MG, Podkaminer K, Yang S, Baker JO, Donohoe BS, Wilson C, Klingeman DM, Olson DG, Decker SR, Giannone RJ, Hettich RL, Brown SD, Lynd LR, Bayer EA, Himmel ME, Bomble YJ (2016) Dramatic performance of Clostridium thermocellum explained by its wide range of cellulase modalities. Scie Adv 2:e1501254. https://doi.org/10.1126/sciadv.1501254
Acknowledgements
Funding was provided by The BioEnergy Science (BESC) and The Center for Bioenergy Innovation (CBI), U.S. Department of Energy Bioenergy Research Centers supported by the Office of Biological and Environmental Research in the DOE Office of Science. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the U.S. DOE under contract DE-AC05-00OR22725. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This manuscript has been authored by UT-Battelle, LLC, under Contract No. DE-AC0500OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for the United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Papanek, B., O’Dell, K.B., Manga, P. et al. Transcriptomic and proteomic changes from medium supplementation and strain evolution in high-yielding Clostridium thermocellum strains. J Ind Microbiol Biotechnol 45, 1007–1015 (2018). https://doi.org/10.1007/s10295-018-2073-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-018-2073-x