
Abstract
Pseudomonas putida is a promising bacterial host for producing natural products, such as polyketides and nonribosomal peptides. In these types of projects, researchers need a genetic toolbox consisting of plasmids, characterized promoters, and techniques for rapidly editing the genome. Past reports described constitutive promoter libraries, a suite of broad host range plasmids that replicate in P. putida, and genome-editing methods. To augment those tools, we have characterized a set of inducible promoters and discovered that IPTG-inducible promoter systems have poor dynamic range due to overexpression of the LacI repressor. By replacing the promoter driving lacI expression with weaker promoters, we increased the fold induction of an IPTG-inducible promoter in P. putida KT2440 to 80-fold. Upon discovering that gene expression from a plasmid was unpredictable when using a high-copy mutant of the BBR1 origin, we determined the copy numbers of several broad host range origins and found that plasmid copy numbers are significantly higher in P. putida KT2440 than in the synthetic biology workhorse, Escherichia coli. Lastly, we developed a λRed/Cas9 recombineering method in P. putida KT2440 using the genetic tools that we characterized. This method enabled the creation of scarless mutations without the need for performing classic two-step integration and marker removal protocols that depend on selection and counterselection genes. With the method, we generated four scarless deletions, three of which we were unable to create using a previously established genome-editing technique.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pseudomonas putida is being developed into a prominent metabolic engineering chassis for industrial biotechnology applications [53]. P. putida naturally has a relatively high guanine–cytosine (GC) content and is capable of expressing complex biosynthetic clusters, such as polyketide synthases and nonribosomal peptide synthetases, making it an ideal host for the production of secondary metabolites derived from GC-rich bacteria [42, 54]. In addition to its production capabilities, P. putida is generally recognized as safe (GRAS)-certified with a relatively high tolerance toward industrial solvents. It has a completely sequenced genome, allowing it to be genetically tractable [52]. For P. putida to reach its potential as a major chassis for industrial chemical production, it requires a robust set of synthetic biology tools to allow for genetic manipulations and modulations of protein expression.
The main synthetic biology tools needed to engineer P. putida include a reliable set of promoters, both constitutive and inducible, a set of origin of replications that can allow for stable plasmid maintenance, and robust and efficient genome-editing techniques [15]. A few constitutive promoter libraries have been characterized in P. putida for chromosomal expression, with one study reporting a dynamic range around three orders of magnitude [69] and a second study finding a 72-fold range of expression [18], demonstrating that a wide range of expression on the chromosome can be achieved.
A variety of inducible promoters natively found in Pseudomonas species have demonstrated in P. putida relatively high levels of expression upon induction, including Pm induced with 3-methylbenzoate [11, 43], Psal induced with salicylate [11, 43], and PalkB induced with dicyclopropylketone [11, 56]. Other heterologous inducible systems have been tested in P. putida with varying success. Induction systems using rhamnose (PrhaB; [11, 30]), arabinose (ParaB; [11, 19]), methyl ethyl ketone (PmekA; [11, 19]), and mannitol (PmtlE; [25]) demonstrated induction levels comparable to the native systems, though basal level expression varied, with PmtlE exhibiting higher levels of basal expression and ParaB acting as a tightly regulated system. The more commonly used Escherichia coli induction systems (Ptet and Plac variants) have also been tested in P. putida, but with varying degrees of success. One study demonstrated 38-fold induction with the Ptet system using four times the reported maximum necessary anhydrotetracycline (aTc) concentration for E. coli [19, 38], and a second study reported using Ptet with tetracycline as the inducer for tubulysin production but did not report quantitative protein expression levels, making it hard to judge the efficacy of Ptet in P. putida [12]. The dynamic range was generally low for systems involving the lac operon, with Ptrc having only a fourfold change in expression upon induction [40]. PlacUV5 was seen to have induced fluorescence levels within the error for the uninduced samples; however, they did report that altering the ribosome binding site (RBS) and 5′ untranslated region (5′ UTR) for PlacUV5 caused a 26-fold increase in expression [11].
Many of the standard plasmid origins commonly used in E. coli are narrow host range origins incapable of replication in P. putida; so a variety of broad host range (BHR) origins have been developed [28, 34, 35]. The RK2 origin is a member of the IncP incompatibility group and requires an origin of replication sequence and a replication initiation protein encoded by trfA to function [41]. It has been shown to be stably maintained in P. putida and has been measured to be a low-copy plasmid in E. coli [9]. RSF1010 is a high-copy BHR origin routinely used for plasmid maintenance in P. putida [5, 51]. It is part of the IncQ incompatibility group, though it requires three genes to successfully replicate (repA, repB, repC), making it a larger origin than RK2 [28, 35]. pBBR1 is an interesting BHR origin that does not seem to belong to any of the standard incompatibility groups and only requires a single gene (rep) for replication, allowing it to be one of the smaller BHR origins [2, 65]. Even with its small size, pBBR1 has been shown to replicate in P. putida and is generally considered a low- to medium-copy plasmid, though the copy number can be altered by mutations in the rep gene [58, 60, 67]. There are also origins that have been isolated from Pseudomonas-specific plasmids (pR01600, pVS1, pNI10), but they must be used in conjunction with other host-specific origins to generate shuttle vectors for P. putida [15, 26].
There are a multitude of techniques for editing the genome of P. putida. Early methods for gene deletions retained the selection marker on the chromosome, limiting the overall number of genomic mutations possible in one strain [63]. To combat this issue, site-specific recombinases such as Cre-loxP have been developed for use in Gram-negative bacteria to allow for efficient removal of the selectable marker [24, 47]. Cre-loxP has been used to efficiently create gene deletions in P. putida KT2440 when paired with the λRed recombinases from the λ bacteriophage, but deletions generated with the Cre-loxP system leaves a loxP scar that can affect future recombination events, also limiting the total number of gene deletions [44].
Markerless and scarless gene-editing methods remove any selectable markers or genomic scars, but they require the use of a counterselection system. In many Gram-negative bacteria, sucrose sensitivity can be conferred by the sacB gene thereby allowing it to be used as a counterselection marker; however, sacB is known to have a high mutation rate that can cause an increase in the false positive rate of mutants [23, 62]. A counterselection for scarless gene deletions based on the antimetabolite 5-fluorouracil was developed, but this technique requires a strain containing the seemingly innocuous deletion of upp, the gene-encoding uracil phosphoribosyltransferase, for functionality [20]. Additionally, most of these methods involve the integration of a suicide plasmid into the chromosome through a single-crossover recombination. This co-integrate can either resolve back to the wild-type sequence or to the desired knockout, resulting in a theoretical efficiency of 50%.
A counterselection based on endonuclease I-SceI allows the use of wild-type P. putida for scarless modifications, but this method also selects for the resolution of an integrated suicide plasmid [46]. Another recently published method found a way to improve the transformation efficiency of sequences integrated into the genome using a serine recombinase system, but this requires the presence of the bacteriophage integrase on the genome and limits its overall utility in genome editing [18].
CRISPR/Cas systems have revolutionized genome editing by providing a fully programmable system, the most common examples using DNA cleavage by the Cas9 nuclease from Streptococcus pyogenes as the counterselection [45]. Cas9 uses a single guide RNA (sgRNA) to target virtually any DNA sequence [32] and has been used successfully for genome editing in both Gram-negative and Gram-positive bacteria [14, 31, 55]. CRISPR/Cas9 cleavage has recently been demonstrated for enhancing the efficiency of single-stranded DNA recombineering in P. putida [4].
Here, we report our characterization of some of the common synthetic biology tools in P. putida. We have tested commonly used E. coli induction systems (Ptrc, PLlacO-I, Ptet and ParaB) in P. putida to determine induction curves and maximum fold induction possible. For the Ptrc and PLlacO-1 promoter systems, we adjusted the expression of lacI in an attempt to improve the fold induction for those systems. We also quantified the plasmid copy number of the BHR origins RK2, pBBR1, and RSF1010 and highlighted the differences in copy number between E. coli and P. putida. Finally, we are reporting a Cas9-assisted recombineering system for P. putida with genome-editing efficiencies approaching 100%.
Materials and methods
Plasmids, bacterial strains, and growth conditions
The plasmids and bacterial strains used in this study are shown in Table S1. E. coli MG1655 and P. putida KT2440 were grown in LB medium at 37 and 30 °C, respectively. LB medium was supplemented with kanamycin (50 µg/mL, Kan50), gentamicin (35 µg/mL, Gent35), tetracyline (10 µg/mL, Tet10 for E. coli; 25 µg/mL, Tet25 for P. putida), or 5-FU (20 µg/mL). 5-FU was purchased from Sigma-Aldrich (F6627-1G).
Induction curves in P. putida
Pseudomonas putida KT2440 strains containing GFPuv expression plasmids were grown overnight in LB Kan50. These overnight strains were inoculated in 5 mL LB Kan50 supplemented with varying amounts of inducer: IPTG (0–2.5 mM), anhydrotetracycline (0–400 ng/mL), and l-arabinose (0–2% w/v). The optical density at 600 nm and fluorescence (excitation: 400 nm, emission: 510 nm) of samples diluted 1:10 in fresh LB media were measured using a Tecan Infinite M1000 plate reader. Analytical flow cytometry was used to measure the fluorescence of cells induced for GFPuv expression. Samples were washed with TBS buffer and diluted 1:10 in fresh TBS buffer before analysis in a BD FACSCalibur. Fluorescence intensities were quantified using the Flowing Software package.
Quantifying plasmid copy number
The copy numbers of five different broad host range origins were quantified in E. coli MG1655 and P. putida KT2440 using quantitative PCR. Individual colonies of P. putida KT2440 strains containing a plasmid were inoculated in LB Kan50 and grown overnight in biological triplicate. Once the cultures reached stationary phase, their genomic and total DNA was extracted using phenol:chloroform:isoamyl alcohol as reported previously, with a few modifications [36, 38]. 1 mL of culture was resuspended in 400 µL 50 mM Tris/50 mM EDTA, pH 8. Cells were lysed by the addition of 8 µL 50 mg/mL lysozyme, followed by incubation at 37 ºC for 30 min. Lysis was continued by adding 4 µL 10% SDS and 8 µL 20 mg/mL proteinase K solution, mixing the samples with a 22-gauge needle, and incubating at 50 ºC for 30 min. Proteinase K was then heat inactivated by incubating the sample at 75 ºC for 10 min, and RNA was digested by adding 2 µL of 10 mg/mL RNase A solution and incubating at 37 ºC for 30 min. The DNA was extracted by mixing the samples with 425 µL of 25:24:1 phenol:chloroform:isoamyl alcohol, vortexing vigorously for 1 min, and letting the samples sit at room temperature for a few minutes. The samples were centrifuged at 14,000g for 5 min at 4 ºC. The upper aqueous phase was transferred to a new tube with a wide-opening pipet tip. DNA extraction was continued by adding 400 µL of chloroform and vortexing and centrifuging the samples as before. The upper aqueous phase was transferred to a new tube. The DNA was further purified by precipitation with 1 volume isopropanol, centrifugation of the samples at maximum speed for 30 min at 4 ºC, washing with 500 µL of 70% ethanol, and rehydrating the DNA by incubating the samples in 200 µL of nuclease-free water at 65 ºC for 1 h.
For determining plasmid copy numbers per cell, individual colonies of E. coli MG1655 and P. putida KT2440 strains containing a plasmid were inoculated in LB Kan50 and grown overnight in biological triplicate. Once the cultures reached the stationary phase, they were diluted in distilled/deionized water to a concentration of 1000 cells/µL (using a conversion factor OD 0.4 = 105 cells/µL).
Quantitative PCR was performed in a 10 µL mixture containing 1X SYBR Green PCR Master Mix (Bio-Rad), 150 nM forward primer, 150 nM reverse primer, and 1 µL of sample. Primers used to determine template concentration were specific to rrsA (for E. coli), lvaC (for P. putida), and gfpuv (for plasmid DNA). Each sample was prepared in technical duplicate. The standard curve for chromosome concentration was made with a dilution series of a P. putida KT2440 genomic DNA extraction from 105 to 1 chromosomes/µL, the standard curve for cell concentration was made with a dilution series of wild-type cells from 104 to 1 cell/µL, and the standard curve for plasmid concentration was made with a dilution series of purified plasmid DNA from 2 × 105 to 20 plasmids/µL. Plasmid and genomic DNA concentrations were measured using the Qubit 3.0 Fluorometer. The reactions were prepared on an AriaMx skirted 96-well plate (Agilent Technologies) and the plate was sealed with an adhesive cover (Bio-Rad). Reactions were run on an AriaMx Real-Time PCR System (Agilent Technologies) using the following cycling conditions: 3 min at 95 ºC, followed by 35 cycles of 15 s at 95 ºC, 30 s at 55 ºC, and 1 min at 72 ºC. Melt curves were generated by increasing the temperature from 55 to 95 ºC using increments of 5 ºC every 5 s. After each run, Cq values were exported to Excel. Technical duplicates with a standard deviation in Cq value greater than 0.3 were not used for analysis. Standard curves were constructed by plotting Cq values vs. the log of chromosome/cell/plasmid concentration. Each standard curve used four to five data points and had an R2 value of at least 0.99. The plasmid copy number was calculated by determining the chromosome/cell/plasmid concentration in each sample using the standard curves and then dividing the plasmid concentration by the chromosome/cell concentration.
Genome editing in P. putida
For the two-step λRed/Cas9 recombineering protocol, P. putida KT2440 containing pRK2-Cas9Red was transformed with pJOE by electroporation and selected on LB Gent, Kan. One of the transformants was inoculated in LB Gent35, Kan50 and grown overnight at 30 ºC. Once the cells reached stationary phase, the λRed genes were induced with 0.5% l-arabinose for 15 min. These cultures were used to prepare electrocompetent cells by washing twice with 10% glycerol and resuspending in 100 µL 10% glycerol for every 1 mL of culture. These cells were transformed with ~ 100 ng pgRNA by electroporation and allowed to recover in 1 mL LB for 2 h at 30 ºC. The recovered cells were selected on LB Gent35, Tet25. For the one-step protocol, P. putida KT2440 containing pCas9 was used to prepare electrocompetent cells in the same way as the two-step protocol. These cells were transformed with ~ 100 ng pgRNA and ~ 500 ng pJOE by electroporation and recovered cells were selected on LB Gent35, Tet25. Genome editing with the 5-FU counterselection was completed as described in Graf and Altenbuchner (2011). Transformants from all three methods were screened for the desired knockout using colony PCR with primers flanking the gene of interest. All positive hits were later screened after re-streaking for isolated colonies with a secondary colony PCR to check for the presence of wild type. Plasmids were cured from P. putida by growing the cells overnight in LB media without antibiotics and plating on LB agar. Single colonies were screened for loss of antibiotic resistance.
Results
Inducible gene expression in P. putida
The Plac, Ptet, and ParaB promoter systems have been used extensively in E. coli and other bacteria [38], but they are not well characterized for gene expression in P. putida. We investigated gene expression from the following promoter systems from the BglBrick vector database: two Plac promoters (1k and 6k), one Ptet promoter (2k), and one ParaB promoter (8k). The Plac promoter systems use the Ptrc (1k) and the PLlacO-1 (6k) promoters. The 1k promoter system also differs from the 6k system, in that lacI is expressed in the same direction as the gene of interest (Fig. 1a). The Ptrc promoter is also a stronger promoter than PLlacO-1 [38]. We identified which promoter systems had high expression levels at maximum induction in P. putida and characterized them by collecting induction curves. We collected the data for these promoter systems after 24 h of growth using the wild-type BBR1 origin and the gfpuv gene as a fluorescent reporter. The BglBrick vector database uses a high-copy mutant of the BBR1 origin, referred to here as BBR1-UP, but our previous work demonstrated that gene expression from plasmids with this origin in P. putida was unreliable, especially when using strong promoters (Figure S1). P. putida cells replicating plasmids with BBR1-UP also had a growth defect (Figure S2).

IPTG-inducible promoter systems from the BglBrick vector database. a Genetic structure of the 1k and 6k promoter systems, highlighting the promoters used and the direction of lacI expression. b Sequences of PlacIq and the three Anderson promoters used to modify the 1k and 6k promoter systems. The − 10 and − 35 motifs are highlighted in red. (Asterisk) Relative activity is the promoter strength in E. coli with respect to the reference promoter in the Anderson promoter library, J23100 (color figure online)
Of the Plac promoters, the 1k system had the highest expression levels at maximum induction, but at high inducer concentrations, gene expression began to decrease (Fig. 2a). The 6k promoter system had extremely poor induction, but PLlacO-1 produces an easily detectable amount of GFPuv in the absence of lacI expression (Fig. 3b). We hypothesized that lacI expression is too high in P. putida for the 6k system and typical levels of inducer cannot de-repress the promoter. We confirmed that the promoter driving lacI expression, PlacIq, is much stronger than a sample of promoters from the Anderson promoter library [1] (Figure S3), and we then modified the 1k and 6k systems by switching PlacIq with these weaker constitutive promoters (Fig. 1b). Gene expression at maximum induction for the modified 1k system was almost double that of the original system and had an 80-fold induction (Figs. 2a, 3a). Gene expression also maintained a steady plateau at high inducer concentrations (Fig. 2a). Increasing expression levels at maximum induction for the 6k system required one of the weakest promoters in the Anderson library, but basal expression increased because lacI expression was too low (Fig. 3b).

Induction curves for BglBrick promoter systems in P. putida. a Induction curves for 1k promoter system with PlacIq and J23107 promoters expressing lacI. b Induction curve for 8k system. Error bars represent one standard deviation. Fluorescence values are adjusted so that the empty vector control has a value of 0

Improving expression at maximum induction for 1k and 6k promoter systems. a Fluorescence for modified 1k promoter systems. b Fluorescence for modified 6k promoter systems. The samples are listed in order of decreasing lacI expression, from left to right. Samples 1k-con and 6k-con are constitutive versions of the 1k and 6k systems, respectively. Error bars represent one standard deviation. Fluorescence values are adjusted so that the empty vector control has a value of 0
Of the remaining promoter systems, the Ptet system did not appear to be functional and induction was detected from the ParaB system (Fig. 2b, Figure S5). The ParaB promoter system provided titratable gene expression over a wide range of l-arabinose concentrations and had a 120-fold induction. However, maximum induction for this promoter system required up to 2% w/v l-arabinose instead of the 0.2% w/v required in E. coli. P. putida does not have an annotated transporter for l-arabinose [49], so we tested the effect of an arabinose transporter, AraE, on protein expression in P. putida cells expressing a fluorescent protein under ParaB with or without expression of araE from a weak promoter (Figure S4a). When araE was constitutively expressed, maximum induction of the ParaB promoter was possible with lower inducer concentrations. Furthermore, P. putida demonstrated homogeneous expression of fluorescent protein as judged by flow cytometry (Figure S4b).
Copy number of broad host range origins
During our previous work using the BBR1-UP origin, we found that it had poor gene expression and plasmid stability in P. putida. This work also resulted in the discovery of BBR1-B5, a mutant of BBR1-UP with an early stop codon in the rep gene, and we found that gene expression was more consistent when using the BBR1-B5 mutant and other BHR origins (Figure S1). We hypothesized that this mutation lowered the copy number of BBR1-UP, resulting in improved reliability in gene expression. We determined the copy number of five BHR origins in E. coli MG1655 and P. putida KT2440 using quantitative PCR (Table 1 and Table S2). Three of the origins were variants of BBR1: wild-type BBR1, BBR1-UP, and BBR1-B5. We also included the origins RK2 and RSF1010 for comparison. For determining plasmid concentration in quantitative PCR, primers targeted the gene gfpuv. LvaC, which is located approximately 3,180,000 bp from the chromosome’s replication origin, was the target for determining chromosome concentration. For all origins, the copy numbers were one order of magnitude higher in P. putida than in E. coli [17, 38, 57]. Both the BBR1 and RK2 origins, which are considered low-copy origins in E. coli, have around 30 copies per chromosomal equivalent in P. putida. The relative copy numbers of the origins are similar between E. coli and P. putida, except for RSF1010, which has a copy number similar to that of BBR1-UP in P. putida. We confirmed that the BBR1-B5 variant has a reduced copy number, which may explain the improved reliability in gene expression when using this origin.
λRed/Cas9 recombineering in P. putida
Our laboratory has had varying success generating knockouts in P. putida using existing methods for genomic deletions. Several knockouts were never constructed due to extremely low editing efficiency (Table S3). To remedy this issue, we developed a λRed/Cas9 recombineering protocol for generating genomic deletions in P. putida. This recombineering method uses an RK2-based plasmid that expresses cas9 from a constitutive, weak promoter (pCas9). The λRed recombinases increase the efficiency of homologous recombination in P. putida [44], so we also included the αβγ operon expressed by the inducible ParaB promoter. While optimizing this system, we attempted Cas9 recombineering without expression of the αβγ operon, but these experiments did not yield any colonies (data not shown). The sgRNA is expressed constitutively from a second plasmid (pgRNAtet), which uses the high-copy BBR1-UP origin. The sgRNA is designed to target the sequence of the chromosome that will be removed upon successful generation of the knockout. To reduce the chances of off-target effects from Cas9/sgRNA expression, we used the CasOT off-target searching tool to identify any off-target sites for potential sgRNAs [68]. The repair template that integrates into the chromosome to generate the knockout is located on a suicide vector (pJOE) [20]. We designed the repair templates for each knockout so that there is 500–1000 bp of homology on either end of the gene of interest and so that the majority of the gene is removed except the start codon and the last 10–20 codons.
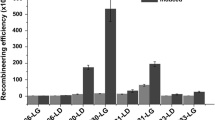
To generate each knockout, we first integrated pJOE into the P. putida KT2440 chromosome via electroporation into a strain replicating pCas9 (Fig. 4a). We used the resulting transformants to prepare electrocompetent cells, during which we induced λRed expression with l-arabinose. Upon introduction of the corresponding pgRNAtet via electroporation, the Cas9/sgRNA complex creates a double-stranded break in the chromosome and the λRed proteins repair the chromosome via homologous recombination. We used this two-step procedure to generate four knockouts in P. putida KT2440 with efficiencies around 85–100% (Table 2). After confirming each knockout, we cured out pCas9 and pgRNAtet by growing liquid cultures overnight in LB media with no antibiotics and plating the cells on LB agar. We screened individual colonies for loss of antibiotic resistance and found that pgRNAtet was easily cured out, but the majority of cells maintained pCas9.

Design and strategy for two-step and one-step λRed/Cas9 recombineering in P. putida KT2440. a The two-step protocol involves a chromosomal integration of pJOE, which carries the repair template for the desired knockout, followed by transformation of the corresponding pgRNA. b The one-step protocol involves a co-transformation of pJOE and pgRNA, where pJOE is used to repair the double-stranded break created by Cas9/sgRNA
To streamline this method, we attempted to generate knockouts with a one-step protocol that involved a co-transformation of pJOE and pgRNAtet (Fig. 4b). Unsurprisingly, the number of transformants and the overall editing efficiency decreased for all four knockouts (Table 2). The rate of detection for the knockout in the initial colony PCR screen was similar to that of the two-step protocol, but after streaking the transformants on selective media for single colonies, we found that most samples lost the designed deletion as well as the wild-type sequence (Figure S7).
Discussion
The inducible promoter systems that we tested are native to E. coli [7, 48, 61], so it is not surprising that they behave differently in P. putida, an organism that does not catabolize l-arabinose or lactose [22, 49]. Expression from ParaB in E. coli results in a mixed population at low inducer concentrations because of an “autocatalytic” induction mechanism involving AraE [64]. Constitutive expression of araE in E. coli enables homogeneous expression from ParaB [33]. Adapting this strategy to P. putida allowed maximum induction at l-arabinose concentrations lower than 2% (Figure S4a). That said, the concentrations needed for maximum induction were less than 0.01% w/v in the presence of araE expression, making it more difficult to titrate gene expression from ParaB and suggesting that araE expression is too high.. Optimizing araE expression may require lowering the gene copy via chromosomal integration or using an alternative transporter with a lower affinity to l-arabinose [50]. IPTG transport is not inhibitory to inducing Plac promoters in P. putida because maximum induction can be easily achieved using concentrations around 1 mM. For the 1k and 6k promoter systems, high expression levels of lacI from the PlacIq promoter reduced gene expression at maximum induction compared to the modified systems. The steady decrease in expression in the induction curve for the 1k system at high inducer concentrations suggests that the reduced expression was due to overproduction of protein, which is similar to the low gene expression observed from strong promoters on a high-copy plasmid (Fig. 2a, Figure S1a). Increasing gene expression from the 6k system required lower lacI expression, but lacI expression for the 6k system must be further optimized so that basal (a.k.a. “leaky”) expression is lower. We did not detect any induction from the Ptet/aTc inducible promoter system (Figure S5). Reducing expression of the TetR repressor could be a successful strategy for improving induction from this promoter system in P. putida. Other reports have shown considerable induction from the Ptet promoter [19, 38], so this system requires further investigation to determine its utility in P. putida. RBS and 5′ UTR sequences also appear to be important for gene expression in P. putida, and differences in these sequences may explain the variability of Ptet in different reports [53, 54].
It is important to consider gene copy number when expressing heterologous genes in bacteria. Gene copy number in E. coli can range from one on bacterial artificial chromosomes (BACs) to 500 on high-copy pUC vectors [29, 37]. Protein production from high-copy plasmids can lead to a metabolic burden and reduce cell growth rate [8], so a complete genetic toolbox for any organism should have low-copy options available. Our quantitative PCR results show that none of the origins tested can be considered low-copy. The inducible promoter systems encoded on BBR1-based plasmids had poor induction due to overexpression of the transcription factors. Lowering the copy number for these promoter systems may improve gene expression by reducing the intracellular concentration of the transcription factors. The copy number of RK2 is dependent on intracellular concentrations of its replication protein, TrfA, at low concentrations, so generating low-copy BHR origins in P. putida may be possible by lowering expression of the replication proteins [16]. Pseudomonas-specific origins that have been shown to be low-copy in other pseudomonads may also have similar properties in P. putida [27]. A guaranteed option for low-copy heterologous gene expression in P. putida is through chromosomal integrations. Either the transcription factor or the entire promoter system could be encoded on the chromosome, depending on the promoter strength. The BBR1 and RK2 origins offer the lowest plasmid copy number of BHR origins commonly used in P. putida, so the probability of overproducing proteins could be limited by using these origins with weak promoters and a single gene or small operon. Even though BBR1-UP leads to unreliable protein production, the efficacy of Cas9/sgRNA activity in our λRed/Cas9 recombineering protocol shows that this origin can be used to reliably express sgRNAs and potentially other non-protein gene products.
The near-100% editing efficiency of Cas9-assisted recombineering in P. putida relies on the counterselection provided by the Cas9/sgRNA complex and the presence of the λRed proteins to facilitate homologous recombination. The most efficient techniques for editing the genome of P. putida involve a single-crossover recombination event between the chromosome and a suicide vector [20, 46]. The counterselections for these methods do not select against the removal of the wild-type sequence, but rather the removal of the suicide vector that can recombine back to the wild-type sequence, resulting in a theoretical editing efficiency of 50%. In practice, deletions may have efficiencies well below 10% and are not reliably identified in a low-throughput genetic screen (Table S3). Existing methods rely on native recombination pathways in P. putida, and therefore they require overnight incubations to resolve the suicide vector. If a knockout causes a growth defect, then any cells that maintain the wild-type sequence will take up a larger percentage of the population as cells replicate, further decreasing the editing efficiency. Designing a sgRNA to target the wild-type sequence for a double-stranded break prevents wild-type cells from outgrowing knockout mutants, providing a theoretical editing efficiency of 100%. The λRed genes can also mediate homologous recombination more rapidly than P. putida’s native recombination pathways, so the cells can resolve the suicide vector from the chromosome within a short 2-h recovery rather than an overnight incubation. That said, the lower editing efficiencies of the one-step protocol suggest that the λRed proteins can facilitate other recombination events in absence of the repair template. These events persist because P. putida is polyploid, and a correctly edited copy of the chromosome can coexist with other incorrect copies until they segregate toward a common DNA sequence. If the repair template was only incorporated into a fraction of the copies of the chromosome, then it is unlikely to survive segregation. This problem does not occur for the two-step protocol because the repair template is already incorporated into every copy of the chromosome after transformation of the pJOE suicide plasmid.
Due to the versatility of Cas9 and BHR origins, this recombineering method could be used to generate deletions in other pseudomonads. The genome-editing technique based on the I-SceI endonuclease developed by de Lorenzo and colleagues originally for P. putida was also used successfully to generate deletions in P. syringae and P. fluorescens [46]. Homologous recombination with the λRed proteins and other related recombination systems has also been demonstrated in multiple pseudomonads [3, 39, 66]. There are several Cas9-assisted recombineering systems available for E. coli that could be adapted for editing the genomes of other Gram-negative bacteria by replacing the E. coli plasmid origins with BHR origins [6, 59].
We demonstrated this technique’s utility in generating knockouts, but it could easily be adapted for integrating heterologous DNA onto the chromosome by including the sequence of interest in the repair template between the regions of homology. Altenbuchner and colleagues integrated a pathway for vanillin production onto the chromosome of P. putida using the 5-FU counterselection developed originally for gene deletions [20, 21]. There are several efficient chromosomal integration systems developed for P. putida [18, 69]. However, these methods are site specific, so they cannot be used to integrate pathways onto multiple loci or modify endogenous pathways. The latter feature is especially necessary for activating cryptic gene clusters in pseudomonads by modifying regulatory elements directly on the chromosome [10, 13].
The results described here establish a common set of genetic tools for use in P. putida. The Plac family of promoters and the ParaB promoter are commonly used in E. coli, but they behave considerably differently in P. putida. Gene expression from the ParaB promoter is the most similar between E. coli and P. putida, but maximum induction in P. putida requires an order of magnitude higher concentration of inducer than in E. coli. Reducing the level of expression of the LacI repressor improved induction from IPTG-inducible promoters, and this strategy could be used to improve other promoter systems with a poor fold induction, such as the Ptet promoter. However, other factors effecting gene expression, such as RBS and 5′ UTR sequence, should also be considered when optimizing gene expression in P. putida. The copy numbers of BHR origins are significantly higher in P. putida, which leads to plasmid instability and unreliable protein production from constitutive and inducible promoters. This fact limits the availability of plasmid-based genetic tools for P. putida; therefore, chromosomal integration should be considered when expressing heterologous genes for its relatively high stability and lower copy number. Taking advantage of CRISPR/Cas9 for editing the genome allows for much more efficient strain development over alternative methods. Shown here as a tool for generating knockouts, λRed/Cas9 recombineering can also be adapted for introducing heterologous genes virtually anywhere on the chromosome or modifying endogenous pathways, providing an alternative platform for metabolic engineering when plasmid-based gene expression may not be optimal.
References
Anderson J (2006) Anderson promoter library registry of standard biological parts. http://partsregistry.org/Promoters/Catalog/Anderson. Accessed 1 Sept 2017
Antoine R, Locht C (1992) Isolation and molecular characterization of a novel broad-host-range plasmid from Bordetella bronchiseptica with sequence similarities to plasmids from gram-positive organisms. Mol Microbiol 6:1785–1799. https://doi.org/10.1111/j.1365-2958.1992.tb01351.x
Aparicio T, Jensen SI, Nielsen AT et al (2016) The Ssr protein (T1E_1405) from Pseudomonas putida DOT-T1E enables oligonucleotide-based recombineering in platform strain P. putida EM42. Biotechnol J 11:1309–1319. https://doi.org/10.1002/biot.201600317
Aparicio T, de Lorenzo V, Martínez-García E (2017) CRISPR/Cas9-based counterselection boosts recombineering efficiency in Pseudomonas putida. Biotechnol J. https://doi.org/10.1002/biot.201700161
Bagdasarian MM, Lurz R, Rückert B et al (1981) Specific-purpose plasmid cloning vectors II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene 16:237–247. https://doi.org/10.1016/0378-1119(81)90080-9
Bassalo MC, Garst AD, Halweg-Edwards AL et al (2016) Rapid and efficient one-step metabolic pathway integration in E. coli. ACS Synth Biol. https://doi.org/10.1021/acssynbio.5b00187
Beckwith JR, Zipser D (1970) The lactose operon. Cold Spring Harbor, New York
Bentley WE, Mirjalili N, Andersen DC et al (1990) Plasmid-encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol Bioeng 35:668–681. https://doi.org/10.1002/bit.260350704
De Bernardez ER, Dhurjati PS (1987) Effect of a broad-host range plasmid on growth dynamics of Escherichia coli and Pseudomonas putida. Biotechnol Bioeng 29:558–565. https://doi.org/10.1002/bit.260290504
Brendel N, Partida-Martinez LP, Scherlach K, Hertweck C (2007) A cryptic PKS–NRPS gene locus in the plant commensal Pseudomonas fluorescens Pf-5 codes for the biosynthesis of an antimitotic rhizoxin complex. Org Biomol Chem 5:2211–2213. https://doi.org/10.1039/b707762a
Calero P, Jensen SI, Nielsen AT (2016) Broad-host-range ProUSER vectors enable fast characterization of inducible promoters and optimization of p-coumaric acid production in Pseudomonas putida KT2440. ACS Synth Biol 5:741–753. https://doi.org/10.1021/acssynbio.6b00081
Chai Y, Shan S, Weissman KJ et al (2012) Heterologous expression and genetic engineering of the tubulysin biosynthetic gene cluster using red/ET recombineering and inactivation mutagenesis. Chem Biol 19:361–371. https://doi.org/10.1016/j.chembiol.2012.01.007
Chiang YM, Chang SL, Oakley BR, Wang CCC (2011) Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms. Curr Opin Chem Biol 15:137–143. https://doi.org/10.1016/j.cbpa.2010.10.011
Cobb RE, Wang Y, Zhao H (2015) High-efficiency multiplex genome editing of streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol 4:723–728. https://doi.org/10.1021/sb500351f
Davison J (2002) Genetic tools for pseudomonads, rhizobia, and other gram-negative bacteria. Biotechniques 32:386–401
Durland RH, Helinski DR (1990) Replication of the broad-host-range plasmid RK2: direct measurement of intracellular concentrations of the essential TrfA replication proteins and their effect on plasmid copy number. J Bacteriol 172:3849–3858. https://doi.org/10.1016/j.mimet.2011.10.007
Durland RH, Toukdarian A, Fang F, Helinski DR (1990) Mutations in the trfA replication gene of the broad-host-range plasmid RK2 result in elevated plasmid copy numbers. J Bacteriol 172:3859–3867
Elmore JR, Furches A, Wolff GN et al (2017) Development of a high efficiency integration system and promoter library for rapid modification of Pseudomonas putida KT2440. Metab Eng Commun 5:1–8. https://doi.org/10.1016/j.meteno.2017.04.001
Graf N, Altenbuchner J (2013) Functional characterization and application of a tightly regulated MekR/P mekA expression system in Escherichia coli and Pseudomonas putida. Appl Microbiol Biotechnol 97:8239–8251. https://doi.org/10.1007/s00253-013-5030-7
Graf N, Altenbuchner J (2011) Development of a method for markerless gene deletion in Pseudomonas putida. Appl Environ Microbiol 77:5549–5552. https://doi.org/10.1128/AEM.05055-11
Graf N, Altenbuchner J (2014) Genetic engineering of Pseudomonas putida KT2440 for rapid and high-yield production of vanillin from ferulic acid. Appl Microbiol Biotechnol 98:137–149. https://doi.org/10.1007/s00253-013-5303-1
Hansen LH, Sørensen SJ, Jensen LB (1997) Chromosomal insertion of the entire Escherichia coli lactose operon, into two strains of Pseudomonas, using a modified mini-Tn5 delivery system. Gene 186:167–173. https://doi.org/10.1016/S0378-1119(96)00688-9
Hashimoto J, Stevenson B, Schmidt TM (2002) Rates and consequences of recombination between ribosomal RNA operons. J Bacteriol 185:966–972. https://doi.org/10.1128/JB.185.3.966
Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. https://doi.org/10.1016/S0378-1119(98)00130-9
Hoffmann J, Altenbuchner J (2015) Functional characterization of the mannitol promoter of Pseudomonas fluorescens DSM 50106 and its application for a mannitol-inducible expression system for Pseudomonas putida KT2440. PLoS One 10:1–22. https://doi.org/10.1371/journal.pone.0133248
Itoh N, Kawanami T, Nitta C et al (2003) Characterization of pNI10 plasmid in Pseudomonas, and the construction of an improved Escherichia and Pseudomonas shuttle vector, pNUK73. Appl Microbiol Biotechnol 61:240–246. https://doi.org/10.1007/s00253-002-1195-1
Itoh Y, Soldati L, Leisinger T, Haas D (1988) Low- and intermediate-copy-number cloning vectors based on the Pseudomonas plasmid pVS1. Antonie Van Leeuwenhoek 54:567–573. https://doi.org/10.1007/BF00588392
Jain A, Srivastava P (2013) Broad host range plasmids. FEMS Microbiol Lett 348:87–96. https://doi.org/10.1111/1574-6968.12241
Jeffrey V, Joachim M (1991) New pUC-derived cloning vectors with different selectable markers and DNA replication origins. Gene 100:189–194. https://doi.org/10.1016/0378-1119(91)90365-I
Jeske M, Altenbuchner J (2010) The Escherichia coli rhamnose promoter rhaPBAD is in Pseudomonas putida KT2440 independent of Crp-cAMP activation. Appl Microbiol Biotechnol 85:1923–1933. https://doi.org/10.1007/s00253-009-2245-8
Jiang W, Bikard D, Cox D et al (2013) RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. https://doi.org/10.1038/nbt.2508
Jinek M, Chylinski K, Fonfara I et al (2012) A programmable dual-RNA—guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–822
Khlebnikov A, Datsenko KA, Skaug T et al (2001) Homogeneous expression of the PBAD promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity araE transporter. Microbiology 147:3241–3247. https://doi.org/10.1099/00221287-147-12-3241
Kovach ME, Elzer PH, Hill DS et al (1995) Four new derivatives of the broad host range cloning vector PBBR1MCS, carrying different antibiotic resistance cassettes. Gene 166:175–176. https://doi.org/10.1089/152045500436104
Kües U, Stahl U (1989) Replication of plasmids in gram-negative bacteria. Microbiol Rev 53:491–516
Lee CL, Ow DSW, Oh SKW (2006) Quantitative real-time polymerase chain reaction for determination of plasmid copy number in bacteria. J Microbiol Methods 65:258–267. https://doi.org/10.1016/j.mimet.2005.07.019
Lee E-C, Yu D, Martinez de Velasco J et al (2001) A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56–65. https://doi.org/10.1006/geno.2000.6451
Lee T, Krupa RA, Zhang F et al (2011) BglBrick vectors and datasheets: a synthetic biology platform for gene expression. J Biol Eng 5:12. https://doi.org/10.1186/1754-1611-5-12
Lesic B, Rahme LG (2008) Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC Mol Biol 9:20. https://doi.org/10.1186/1471-2199-9-20
Lieder S, Nikel PI, de Lorenzo V, Takors R (2015) Genome reduction boosts heterologous gene expression in Pseudomonas putida. Microb Cell Fact 14:23. https://doi.org/10.1186/s12934-015-0207-7
Lin J, Helinski DR (1992) Analysis of mutations in trfA, the replication initiation gene of the broad-host-range plasmid RK2. J Bacteriol 174:4110–4119
Loeschcke A, Thies S (2015) Pseudomonas putida—a versatile host for the production of natural products. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-015-6745-4
de Lorenzo V, Fernández S, Herrero M et al (1993) Engineering of alkyl- and haloaromatic-responsive gene expression with mini-transposons containing regulated promoters of biodegradative pathways of Pseudomonas. Gene 130:41–46. https://doi.org/10.1016/0378-1119(93)90344-3
Luo X, Yang Y, Ling W et al (2016) Pseudomonas putida KT2440 markerless gene deletion using a combination of λ Red recombineering and Cre/loxP site-specific recombination. FEMS Microbiol Lett. https://doi.org/10.1093/femsle/fnw014
Mali P, Esvelt KM, Church GM (2013) Cas9 as a versatile tool for engineering biology. Nat Methods 10:957–963. https://doi.org/10.1038/nmeth.2649
Martínez-García E, de Lorenzo V (2011) Engineering multiple genomic deletions in gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ Microbiol 13:2702–2716. https://doi.org/10.1111/j.1462-2920.2011.02538.x
Marx CJ, Lidstrom ME (2002) Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques 33:1062–1067
McMurry L, Petrucci RE, Levy SB (1980) Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. Proc Natl Acad Sci USA 77:3974–3977. https://doi.org/10.1073/pnas.77.7.3974
Meijnen JP, De Winde JH, Ruijssenaars HJ (2008) Engineering Pseudomonas putida S12 for efficient utilization of d-xylose and l-arabinose. Appl Environ Microbiol 74:5031–5037. https://doi.org/10.1128/AEM.00924-08
Morgan-Kiss RM, Wadler C, Cronan JE (2002) Long-term and homogeneous regulation of the Escherichia coli araBAD promoter by use of a lactose transporter of relaxed specificity. Proc Natl Acad Sci USA 99:7373–7377. https://doi.org/10.1073/pnas.122227599
Nagahari K, Sakaguchi K (1978) RSF1010 plasmid as a potentially useful vector in Pseudomonas species. J Bacteriol 133:1527–1529
Nelson KE, Weinel C, Paulsen IT et al (2002) Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ Microbiol 4:799–808. https://doi.org/10.1046/j.1462-2920.2002.00366.x
Nikel PI, Chavarría M, Danchin A, de Lorenzo V (2016) From dirt to industrial applications: Pseudomonas putida as a synthetic biology chassis for hosting harsh biochemical reactions. Curr Opin Chem Biol 34:20–29. https://doi.org/10.1016/j.cbpa.2016.05.011
Nikel PI, Martínez-García E, de Lorenzo V (2014) Biotechnological domestication of pseudomonads using synthetic biology. Nat Rev Microbiol 12:368–379. https://doi.org/10.1038/nrmicro3253
Oh J-H, van Pijkeren J-P (2014) CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res 42:1–11. https://doi.org/10.1093/nar/gku623
Panke S, De Lorenzo V, Kaiser A, Wubbolts MG (1999) Engineering of a stable whole-cell biocatalyst capable of (S)-styrene oxide formation for continuous two-liquid-phase applications. Appl Environ Microbiol 65:5619–5623
Persson C, Nordström K (1986) Control of replication of the broad host range plasmid RSF1010: the incompatibility determinant consists of directly repeated DNA sequences. Mol Gen Genet 203:189–192. https://doi.org/10.1007/BF00330402
Prior JE, Lynch MD, Gill RT (2010) Broad-host-range vectors for protein expression across gram negative hosts. Biotechnol Bioeng 106:326–332. https://doi.org/10.1002/bit.22695
Pyne ME, Moo-Young M, Chung DA, Chou CP (2015) Coupling the CRISPR/Cas9 system with lambda red recombineering enables simplified chromosomal gene replacement in Escherichia coli. Appl Environ Microbiol 81:5103–5114. https://doi.org/10.1128/AEM.01248-15
Rand JM, Pisithkul T, Clark RL et al (2017) A metabolic pathway for catabolizing levulinic acid in bacteria. Nat Microbiol. https://doi.org/10.1038/s41564-017-0028-z
Rvbetzt K (1981) Regulation of the l-arabinose transport operons in Escherichia coli. J Mol Biol 151:215–227
Schäfer A, Tauch A, Jäger W et al (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. https://doi.org/10.1016/0378-1119(94)90324-7
Schweizer HP, Hoang TT (1995) An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene 158:15–22. https://doi.org/10.1016/0378-1119(95)00055-B
Siegele DA, Hu JC (1997) Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc Natl Acad Sci USA 94:8168–8172. https://doi.org/10.1073/pnas.94.15.8168
Silva-Rocha R, Martínez-García E, Calles B et al (2013) The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res 41:666–675. https://doi.org/10.1093/nar/gks1119
Swingle B, Bao Z, Markel E et al (2010) Recombineering using RecTE from Pseudomonas syringae. Appl Environ Microbiol 76:4960–4968. https://doi.org/10.1128/AEM.00911-10
Tao L, Jackson RE, Cheng Q (2005) Directed evolution of copy number of a broad host range plasmid for metabolic engineering. Metab Eng 7:10–17. https://doi.org/10.1016/j.ymben.2004.05.006
Xiao A, Cheng Z, Kong L et al (2014) CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 30:1180–1182. https://doi.org/10.1093/bioinformatics/btt764
Zobel S, Benedetti I, Eisenbach L et al (2015) Tn7-based device for calibrated heterologous gene expression in Pseudomonas putida. ACS Synth Biol. https://doi.org/10.1021/acssynbio.5b00058
Acknowledgements
This study was supported by research grants from the National Science Foundation (CBET-114678, MCB-1716594) and US-AID (PEER 3-195). T.B.C. and D.K.C. are recipients of NIH Biotechnology Training Program Fellowships (NIGMS 5 T32 GM08349). J.M.R. was supported by an NSF Graduate Research Fellowship (DGE-1256259). S.A.L. is the recipient of a fellowship from the Promega Corporation through the Dane County Youth Apprenticeship Program in Biotechnology. The authors would like to thank Dr. Yalun Arafin and Dr. Fransiskus Ivan for their assistance with the project.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cook, T.B., Rand, J.M., Nurani, W. et al. Genetic tools for reliable gene expression and recombineering in Pseudomonas putida. J Ind Microbiol Biotechnol 45, 517–527 (2018). https://doi.org/10.1007/s10295-017-2001-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-017-2001-5