
Abstract
Single-cell biorefineries are an interesting strategy for using different components of feedstock to produce multiple high-value biochemicals. In this study, a strategy was applied to refine glucose and fatty acid to produce 5-aminolevulinic acid (ALA) and polyhydroxyalkanoates (PHAs). To express the ALA and PHAs dual-production system efficiently and stably, multiple copies of the poly-β-3-hydroxybutyrate (PHB) synthesis operon were integrated into the chromosome of Escherichia coli DH5αΔpoxB. The above strain harboring the ALA C5 synthesis pathway genes hemA and hemL resulted in coproduction of 38.2% PHB (cell dry weight, CDW) and 3.2 g/L extracellular ALA. To explore coproduction of ALA and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV), the PHBV synthetic pathway was also integrated into engineered E. coli and coexpressed with hemA and hemL; cells produced 38.9% PHBV (CDW) with 10.3 mol% 3HV fractions and 3.0 g/L ALA. The coproduction of ALA with PHB and PHBV can improve the utilization of carbon sources and maximize the value derived from the feedstock.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Biorefineries are a fascinating approach to transforming renewable raw materials into separate bio-based process streams and ultimately chemicals and fuels [24]. Coproduction systems are an attractive biorefinery strategy, in which engineered microorganisms are capable of accumulating multiple products. This approach has advantages in balancing in vivo metabolic flux and restoring optimal cell physiology. Therefore, many efforts have been made to develop various coproduction systems to reduce costs and improve production efficiency [20].
5-Aminolevulinic acid (ALA), a non-protein amino acid, is the common precursor of almost all the pyrrole compounds in plants, animals, bacteria and fungi, including vitamin B12, chlorophyll, porphyrins and heme. ALA has attracted much attention for its potential applications in medicine in second-generation photodynamic drugs. In living organisms, there are two major pathways for ALA biosynthesis, named C4 and C5. In the C4 pathway, ALA is formed through the condensation of glycine and succinyl-CoA catalyzed by 5-aminolevulinate synthase. Production of ALA via the C4 pathway, intensely studied in Escherichia coli and Corynebacterium glutamicum, requires the artificial addition of glycine or/and succinyl-CoA [13, 21, 35, 37]. Our previous paper developed a new strategy to produce ALA in recombinant E. coli via the C5 pathway. The C5 pathway utilizes α-ketoglutarate, a C5 intermediate of the tricarboxylic acid (TCA) cycle, as the carbon skeleton. Heterologous, stabilized glutamyl-tRNA reductase HemA from Salmonella arizona was introduced into E. coli and coexpressed with glutamate-1-semialdehyde aminotransferase HemL and an ALA exporter. The recombinant E. coli produced 4.13 g/L ALA in modified minimal medium using glucose as the sole carbon source [12].
In our previous study, we observed acetate accumulation by the ALA-producing E. coli strain DALA. To remove this metabolic imbalance, we designed to introduce polyhydroxyalkanoates (PHAs) biosynthesis pathway into the ALA-producing strain. In E. coli, the main source of acetate is acetyl-CoA; thus, a product using AcCoA as a precursor is needed to prevent acetate accumulation. PHAs are a type of biodegradable and biocompatible polyester accumulated by many microorganisms [3, 9, 29]; they are considered potential “green” alternatives to petrochemical-based plastics for use in drug delivery, agriculture, fibers and consumer products [2, 31]. Poly-β-3-hydroxybutyrate (PHB) is the best-known PHA. The in vivo biosynthesis of PHB only requires three steps using AcCoA as the precursor, and the production of PHB has been intensively studied [15, 18, 28]. Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) is commercially interesting because of its physical properties, including melting point, crystal growth rate, plasticity, and biodegradability [25, 27]. The cost-effective production of PHBV in microorganisms needs an increased propionyl-CoA precursor supply for the 3-hydroxyvalerate (3HV) fraction in PHBV to avoid the need for addition of the second carbon source propionate, which is toxic and expensive [7]. Various strategies have been applied to improve the 3HV fraction content for PHBV synthesis from other carbon sources, including manipulation of the citramalate pathway [34, 36] and branched amino acid (e.g., threonine, isoleucine) pathways [1, 6, 30]. Moreover, to block propionyl-CoA to many other end products pathways, prpC and scpC double deletion was demonstrated effective to inhibit the endogenous propionyl-CoA catabolism [6]. The prpC gene encodes a methylcitrate synthase and catalyzes the conversion of propionyl-CoA to methylcitrate; scpC encodes the propionyl-CoA:succinyl-CoA transferase and catalyzes the reaction converting propionyl-CoA into succinyl-CoA.
In this study, we achieved the coproduction of PHAs and ALA in recombinant E. coli (Fig. 1). The combined production of ALA and PHAs may maintain balanced metabolic flux in the cell, and the accumulation of intracellular PHA has been shown to improve the stress resistance of host cells [4, 33], which thus increases the carbon conversion ratio or/and promotes the yield of both products. Intracellular accumulation of PHA and secretion of ALA will make separation of the two products easily, reducing downstream costs [19]. Single-cell biorefinery can break through the traditional fermentation model. The realization of ALA and PHAs coproduction has important practical significance.

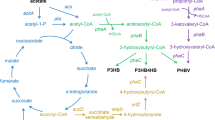
Designed synthetic pathways for 5-aminolevulinic acid (ALA) and polyhydroxyalkanoate (PHA) production constructed in Escherichia coli. Genes and their enzymes: phbA β-ketothiolase; phbB acetoacetyl-CoA reductase; phbC poly-β-3-hydroxybutyrate (PHB) synthase; hemA glutamyl-tRNA reductase from Salmonella arizona; hemL glutamate 1-semialdehyde aminotransferase; thrA* 1034C→T mutated aspartate kinase/homoserine dehydrogenase
Materials and methods
Bacterial strains and plasmids
Table 1 lists all the microorganisms and plasmids used in this study.
Genome integration of PHB operon for construction of E. coli strain XLPHB
The chromosomal integration of multiple copies of the PHB operon was performed using the CIGMC method, which uses flippase for specific recombination at FLP/FRT sites [10]. In brief, E. coli DH5αΔpoxB was used as the starting strain for genome integration. The phaCAB genes cloned from pBHR68 were inserted into the NsiI and XhoI sites of p5TG, resulting in plasmid p5TPHB. p5TCAB was digested with HindIII, and XhoI. pKD4 was used as the template to amplify FRT-Kan, using primers FRT-Kan-F and FRT-Kan-R. The pUC19 vector backbone was obtained by PCR using primers pUC19-F and pUC19-R. Then, the latter three fragments were assembled in vitro by Gibson assembly in proper proportions. After transformation, the recombinant plasmid pAS4 was obtained. pAS4 was cut with EcoRI to obtain the target fragment FRT-kan-P5tac-phaCAB. Then, the fragment was cyclized by T4 ligase at 25 °C for 30 min. pCP20 was transformed into E. coli strain DH5αΔpoxB to construct strain DH5αΔpoxB/pCP20. This strain was inoculated into 5 mL of Luria–Bertani medium (LB; 10 g/L NaCl, 5 g/L yeast extract and 10 g/L tryptone) at 30 °C with shaking at 250 rpm overnight. Then, 1-mL cell cultures were transferred to 50 mL SOB medium (2% tryptone, 0.5% yeast extract, 0.05% NaCl, 2.5 mM KCl and 10 mM MgCl2) and incubated at 30 °C to an optical density at 600 nm (OD600) of 0.5. Cells were then cultivated at 42 °C to induce the expression of FLP recombinase for 20–30 min and harvested for electroporation. The cyclized Kan-P5tac-phaCAB fragments were electrotransformed into the above competent cells and then spread on kanamycin-containing plates overnight. Colony PCR was used to check whether the integration was successful.
The integrated copy number of phaCAB was accurately determined by qPCR. Total cellular mRNA was extracted using an RNeasy Mini Kit (Tiangen) after strains were cultivated in shaken flasks for 3 h. cDNA was synthesized by reverse transcription PCR using random hexamers and Oligo-dT as primers with a Prime Script RT Reagent Kit (Takara). qRT-PCR was carried out using a Light Cycler 480 Real-Time PCR System (Roche) with SYBR Premix Ex Taq™ II (Takara). The measurement of each gene was repeated three times. The gapA gene, of which the expression level is relatively constant, was used as the control gene. DH5αΔpoxB::(P5tac-phbCAB), which had one copy of the P5tac-phaCAB operon in the genome, was used as the control strain.
Genome integration of phbCBB for construction of E. coli strain XL103
The recombinant strain XL103 was also obtained by specific recombination at a FLP/FRT site. The gene bktB, cut from pCBB, was ligated into pAS4 to replace phaA using StuI to obtain the fragment P5tac-phaCBB. Primers (Kan-R6K) F and (Kan-R6K) R were used to amplify the Kan-R6K fragment, which included the kanamycin resistance gene and the narrow-host-range replicon R6K from pKD4. The two fragments were assembled with the Gibson system to obtain the integrated plasmid pQXL. pQXL was transformed into strain QW4, in which the threonine metabolic pathway was overexpressed. Chromosomal integration using pCP20 was performed as described above, and the integrated copy number of phaCBB was determined by qPCR.
Medium and culture conditions
For plasmid construction, strains were cultivated on LB agar plates or in LB broth. SOB medium was used for genome integration. For PHB production, modified M9 medium was used [6]. For coproduction of PHB (or PHBV) and ALA, modified minimal medium was used for fermentation (5 g/L (NH4)2SO4, 3 g/L KH2PO4, 16 g/L Na2HPO4·12H2O, 1 g/L MgSO4·7H2O, 0.01 g/L MnSO4·7H2O and 2 g/L yeast extract). Glucose was used as the sole carbon source and was added as indicated. Ampicillin (100 μg/mL), kanamycin (25 μg/mL), chloramphenicol (25 μg/mL) or spectinomycin (25 μg/mL) was added to provide selective pressure during cultivation or for gene integration selection when necessary. To induce the expression of plasmid-borne genes, isopropyl-β-d-thiogalactopyranoside (IPTG) was added into cultures with a final concentration of 1 mM.
For all shaken flask culture experiments, single colonies were inoculated into 5-mL LB broth and grown at 37 °C overnight. Preculture [2% (v/v)] was inoculated into 300-mL Erlenmeyer flasks containing 50-mL fermentation medium and cultivated at 37 °C with shaking at 200 rpm. When the OD600 reached 0.6–0.8, 1 mM IPTG was added to the culture broth as an inducer. After induction, 40 g/L glucose was supplied as the sole carbon source and cells were cultured for 60 h at 37 °C with shaking at 200 rpm.
Analytical methods
For monitoring microbial growth, OD600 was measured with a spectrophotometer. ALA concentration was analyzed using modified Ehrlich’s reagent [5, 22]. Heme concentration was assayed by a Hemin Colorimetric Assay Kit (Biovision, Catalog K672-100). Metabolites such as acetate were measured using a high-performance liquid chromatography (HPLC) system (Shimadzu, Japan) equipped with an HPX-87H column (300 mm × 7.8 mm; Bio-Rad, USA) and a refractive index detector (Shimadzu RID-10A). Samples analyzed using HPLC were filtered through a 0.22-µm-pore syringe filter. The mobile phase (5 mM H2SO4) was delivered at 0.6 mL/min at 65 °C. An Orion RDO meter (Thermo Fisher Scientific, USA) was used to determine oxygen uptake rates. Each culture (50 mL) was harvested during the exponential growth phase (14–16 h), and the OD600 was recorded. The cells were pelleted by centrifugation and washed once with sterilize phosphate-buffered saline (pH 7.5) and then diluted to 0.5 OD600, and 100 mL of the diluted cell suspension was cultured in medium containing 30 g/L glucose at 37 °C with shaking at 200 rpm. The slopes of curves of the dissolved oxygen (DO) concentration vs time were determined from 70 to 20% air saturation. OURs were determined according to the OD600 of the cell suspension and presented as mg O2 consumed per OD600 biomass per minute [32]. For PHAs detection, cells were harvested by centrifugation at 10,000×g for 10 min. The cell pellets were washed twice with distilled water and then lyophilized for 7 h. Cell dry weights (CDW) were measured after the lyophilization of cell pellets. PHB and PHBV were quantified by gas chromatography (GC).
Before GC analysis, 1 mL chloroform, 850 μL methanol, and 150 μL sulfuric acid (98%, w/w) were added to the weighed cells. The vials were incubated at 100 °C for 1 h. Then, 1 mL water was added to the cool vials. After phase separation, the heavier chloroform phase was transferred to a fresh vial for GC analysis. The GC detection process was performed using a Shimadzu GC2010 (Kyoto, Japan) equipped with an AOC-20i auto injector and a Restek Rtx-5 column. PHAs standard samples were also analyzed by GC according to the above method. The temperature program for PHB and PHBV was: 80 °C for 1 min, ramp to 120 °C at 10 °C/min, ramp to 160 °C at 45 °C/min, hold for 5 min.
Results
Coproduction of PHB and ALA in PHB chromosomally integrated E. coli strain XLPHB using glucose
We selected E. coli DH5αΔpoxB as the basal strain. The poxB gene was knocked out leaving a FRT site in the genome. Thus, the multiplexed PHB synthesis operon could be integrated into the genome using FLP-/FRT-mediated site-specific recombination. After screening, 22 clones were obtained which was integrated into the PHB operon successfully. The integrated copy number of each strain was detected by colony PCR and qPCR (Fig. 2a). Every clone contained 1–3 copies of PHB operon. We chose the strains with three integrated copies for shaken flask fermentation. The seed culture medium was LB, and the fermentation medium was modified M9. The PHB yield of strain XLPHB could reach about 50% of the cell dry weight (CDW) (ranged from 41.4 to 51.3%), which was comparable with a recombinant strain harboring a high-copy-number plasmid carrying the PHB operon [34].

qPCR detection of the copy numbers of phbCAB and phbCBB operons integrated into E. coli strains XLPHB (a) and XL103 (b). The experiments were performed in triplicate, and error bars indicate standard deviation (SD)
To coproduce PHB and ALA, pDAL and pCLRA were transformed into the PHB producing strain E. coli XLPHB; untransformed XLPHB was cultured for comparison. Strain XLPHB accumulated 1.76 g/L PHB, which was about 43.1% of the CDW (Fig. 3). When pDAL was transformed into strain XLPHB, combined production of PHB and ALA was achieved. Strain XLPHB (pDAL) accumulated 1.54 g/L PHB in the cell, accounting for 41.5% of the CDW and 2.5 g/L ALA in the culture medium. When pDAL and pCLRA were cotransformed into XLPHB, the cells accumulated 1.38 g/L of PHB, about 38.2% of the CDW and 3.2 g/L of ALA. DALA-PHB accumulated the highest ALA titer 2.6 g/L at 26 h, while PHB began to rapid accumulate after 48 h, when the nitrogen source was exhaust (Fig. 4). These results demonstrated that we successfully established the distinct pathways for coproducing PHB and ALA in E. coli.

PHB and ALA coproduction in E. coli strain XLPHB harboring different plasmids. The experiments were performed in triplicate, and error bars indicate standard deviation (SD)

Time course profile of a E. coli XLPHB and b DALA-PHB during batch cultivation. Open triangles growth curves, filled circles glucose consumption and filled triangles ALA concentration, columns PHB content. The experiments were performed in triplicate, and error bars indicate standard deviation (SD)
Coproduction of PHBV and ALA in PHBV chromosomally integrated E. coli strain XL103 using glucose
For PHBV pathway genome integration, pQXL was transformed into strain XL102/pCP20, and the recombinant strain E. coli XL103 was obtained by specific recombination under the action of FLP. Twenty-nine clones were screened by qPCR to determine the copy number integrated into the genome (Fig. 2b). Most of the strains had four to six copies of the phaCBB operon, a few integrated one or two copies, while clone XL103-14 incorporated nine copies of the operon. We chose XL103-14 as the base strain for production of PHBV; it accumulated PHBV to about 43% of the CDW.
Previous results showed that overexpression of threonine deaminase gene ilvA from C. glutamicum increased the 3HV fraction in the PHBV copolymer [6]. So, we examined its effect on PHBV production in strain XL103-14 by transforming pBBR-ilvACG. Strains XL103 and XL103 (pBBR-ilvACG) were used to produce PHBV by fermentation in shaken flasks with glucose as the sole carbon source (Fig. 5). Strain XL103 could accumulate a large amount of PHB (> 40% of the CDW), but only a very small amount of 3HV without expressing ilvA. When ilvA was expressed, the recombinant strain accumulated PHBV with elevated 3HV content. The fraction of 3HV was 12.4 mol%, while the total amount of PHBV (by CDW) was slightly decreased compared with XL103 not expressing ilvA. Coproduction of ALA and PHBV was achieved when pDAL and pDAL + pCLRA were transformed into XL103 (pBBR-ilvACG). The ALA production was 2.4 and 3.0 g/L, respectively, while the yields of PHBV were 42.1 and 38.9% with 11.7 and 10.3% 3HV fraction, respectively.

Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) and ALA coproduction in E. coli XL103 harboring different plasmids. The experiments were performed in triplicate, and error bars indicate standard deviation (SD)
Synergy of ALA and PHAs coproduction by maintaining metabolic balance
Coproduction of ALA and PHAs improved the use of the carbon substrate. To investigate if the two coproducts are functionally complementary, some metabolic and physiological parameters of these strains were analyzed (Fig. 6). ALA-producing strain DALA accumulated a large quantity of acetate (7.45 g/L), indicating imbalanced metabolism of the strain. The inactivation of the pyruvate oxidase gene poxB in strains XLPHB and XL103 reduced the acetate secretion by no more than 30%. In the coproducing strains DALA-PHB and DALA-103, acetate accumulation reduced to 0.9 g/L and even lower, indicating that the carbon overflow to acetate was pushed into PHBV synthesis pathway. The intracellular heme concentration and the oxygen uptake rate of engineered strains were also determined. Since heme is an essential iron-containing component of proteins in the electron transport chain and TCA cycle that drives aerobic and anaerobic respiration, its excessive production would influence metabolism. ALA production resulted in excessive accumulation of the downstream product heme, which is increased by almost tenfold compared with that of XLPHB or XL103. The oxygen uptake rate was increased by about 15%. This indicated that excessive heme influenced respiratory efficiency, and further may change the energy generation pathway and metabolic flux of E. coli.

Acetate profile, heme production and oxygen uptake rate in engineered E. coli strains. The experiments were performed in triplicate, and error bars indicate standard deviation (SD)
Discussion
In this study, we achieved the coproduction of different PHAs with ALA. Compared with single product processes, this coproduction strategy can use substrate more effectively. During the coproduction of PHB and ALA, the production of PHB was about 1.54 g/L, and ALA was accumulated > 3 g/L. When the block copolymers PHBV and ALA were coproduced, PHBV accumulation was up to 40% of the CDW and ALA production was about 3 g/L.
Recently, Li et al. reported a coproduction of PHB and ALA in recombinant E. coli by engineering the C4 pathway which condensed succinyl-CoA with glycine by ALA synthase. The results showed a coproduction of 43% PHB (CDW) and 1.6 g/L extracellular ALA [19]. However, this production process required the addition of glycine and succinic acid. Plasmid-dependent overexpression still has some problems, such as genetic instability and lack of repeatability. Previously, high instability was observed when recombinant E. coli accumulated PHB [16]. Therefore, for stable expression, we used a one-step CIGMC method based on FLP recombinase to integrate multiple copies of PHB biosynthesis genes into the chromosome. Stable PHB production was obtained, and the accumulation of second product, ALA, was almost unaffected by the accumulation of PHB.
A PHBV-producing strain was also constructed in our study. E. coli QW4 was genomically modified to overproduce l-threonine by removing the feedback inhibition on aspartokinase I by a site-specific (1034C → T) mutation of thrA and a Ptac promoter substitution [23]. prpC, scpC and poxB were also knocked out in this strain. Thus, QW4 was used as the original strain for PHB operon integration. Then, we achieved PHBV and ALA coproduction for the first time.
Physiological experiments of DALA-PHB and DALA103 with their compared strains revealed decreased acetate accumulation and an increased respiration rate when two products were simultaneously synthesized. PHB accumulation resulted in a reduction in acetate accumulation, saving the carbon source. Moreover, PHB accumulation is beneficial for host resistance [33], which increases the survival rate of the bacteria and contributes to fermentative production. ALA accumulation resulted in excess heme production. Although an excess of this critical cofactor is toxic to bacteria [14, 26], it can lead to the up-regulation of genes involved in respiration chain [38]. The improved respiration rate likely increased the flux of the TCA cycle, which decreased overflow metabolism, including decreased production of acetate. Further, the enhanced respiration rate and the TCA cycle flux likely change energy generation mediated by the electron transport chain. So, our system resulted in functional complementarity of both products and ensured the production of both by the efficient use of carbon and energy.
The single-cell biorefinery proved to be an interesting strategy for efficient-utilization of feedstock and producing high-value ALA and PHAs. It can also take advantage of various components in the biomass and their intermediates to produce multiple products, thereby maximizing the value derived from the feedstock [11]. However, there are also some issues in the ALA and PHAs coproduction process such as competition for carbon and reducing power. Challenges must be overcome before putting these systems into industrial settings. Coproduction processes involving optimized single-cell biorefineries have potential application in the production of value-added commodity chemicals.
References
Aldor IS, Kim SW, Prather KL, Keasling JD (2002) Metabolic engineering of a novel propionate-independent pathway for the production of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) in recombinant Salmonella enterica serovar Typhimurium. Appl Environ Microbiol 68(8):3848–3854
Aldor IS, Keasling JD (2003) Process design for microbial plastic factories: metabolic engineering of polyhydroxyalkanoates. Curr Opin Biotechnol 14(5):475–483
Anderson AJ, Dawes EA (1990) Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol Rev 54(4):450–472
Ayub ND, Pettinari MJ, Ruiz JA, Lopez NI (2004) A polyhydroxybutyrate-producing Pseudomonas sp. isolated from Antarctic environments with high stress resistance. Curr Microbiol 49(3):170–174
Burnham DC (1970) Simple measurement of thermal lensing effects in laser rods. Appl Opt 9(7):1727–1728
Chen Q, Wang Q, Wei G, Liang Q, Qi Q (2011) Production in Escherichia coli of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) with differing monomer compositions from unrelated carbon sources. Appl Environ Microbiol 77(14):4886–4893
Choi JI, Lee SY (1999) High-level production of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) by fed-batch culture of recombinant Escherichia coli. Appl Environ Microbiol 65(10):4363–4368
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97(12):6640–6645
Doi Y, Segawa A, Kawaguchi Y, Kunioka M (1990) Cyclic nature of poly(3-hydroxyalkanoate) metabolism in Alcaligenes eutrophus. FEMS Microbiol Lett 55(1–2):165–169
Gu P, Yang F, Su T, Wang Q, Liang Q, Qi Q (2015) A rapid and reliable strategy for chromosomal integration of gene(s) with multiple copies. Sci Rep 5:9684
Kang Z, Gao C, Wang Q, Liu H, Qi Q (2010) A novel strategy for succinate and polyhydroxybutyrate co-production in Escherichia coli. Bioresour Technol 101(19):7675–7678
Kang Z, Wang Y, Gu P, Wang Q, Qi Q (2011) Engineering Escherichia coli for efficient production of 5-aminolevulinic acid from glucose. Metab Eng 13(5):492–498
Kiatpapan P, Murooka Y (2001) Construction of an expression vector for propionibacteria and its use in production of 5-aminolevulinic acid by Propionibacterium freudenreichii. Appl Microbiol Biotechnol 56(1–2):144–149
Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A et al (2010) A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med 2(51):51ra71
Lee SY, Lee KM, Chan HN, Steinbuchel A (1994) Comparison of recombinant Escherichia coli strains for synthesis and accumulation of poly-(3-hydroxybutyric acid) and morphological changes. Biotechnol Bioeng 44(11):1337–1347
Lee SY, Yim KS, Chang HN, Chang YK (1994) Construction of plasmids, estimation of plasmid stability, and use of stable plasmids for the production of poly(3-hydroxybutyric acid) by recombinant Escherichia coli. J Biotechnol 32(2):203–211
Li M, Wang J, Geng Y, Li Y, Wang Q, Liang Q, Qi Q (2012) A strategy of gene overexpression based on tandem repetitive promoters in Escherichia coli. Microb Cell Fact 11:19
Li R, Chen Q, Wang PG, Qi Q (2007) A novel-designed Escherichia coli for the production of various polyhydroxyalkanoates from inexpensive substrate mixture. Appl Microbiol Biotechnol 75(5):1103–1109
Li T, Guo YY, Qiao GQ, Chen GQ (2016) Microbial synthesis of 5-aminolevulinic acid and its coproduction with polyhydroxybutyrate. ACS Synth Biol 5(11):1264–1274
Liang Q, Qi Q (2014) From a co-production design to an integrated single-cell biorefinery. Biotechnol Adv 32(7):1328–1335
Lin J, Fu W, Cen P (2009) Characterization of 5-aminolevulinate synthase from Agrobacterium radiobacter, screening new inhibitors for 5-aminolevulinate dehydratase from Escherichia coli and their potential use for high 5-aminolevulinate production. Bioresour Technol 100(7):2293–2297
Mauzerall D, Granick S (1956) The occurrence and determination of delta-amino-levulinic acid and porphobilinogen in urine. J Biol Chem 219(1):435–446
Patte J (1996) Biosynthesis of threonine and lysine. In: Escherichia coli and Salmonella: cellular and molecular biology, vol 1. pp 528–541
Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ Jr, Hallett JP, Leak DJ, Liotta CL et al (2006) The path forward for biofuels and biomaterials. Science 311(5760):484–489
Ren Q, Sierro N, Kellerhals M, Kessler B, Witholt B (2000) Properties of engineered poly-3-hydroxyalkanoates produced in recombinant Escherichia coli strains. Appl Environ Microbiol 66(4):1311–1320
Ryter SW, Tyrrell RM (2000) The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med 28(2):289–309
Shang L, Yim SC, Park HG, Chang HN (2004) Sequential feeding of glucose and valerate in a fed-batch culture of Ralstonia eutropha for production of poly(hydroxybutyrate-co-hydroxyvalerate) with high 3-hydroxyvalerate fraction. Biotechnol Prog 20(1):140–144
Slater SC, Voige WH, Dennis DE (1988) Cloning and expression in Escherichia coli of the Alcaligenes eutrophus H16 poly-beta-hydroxybutyrate biosynthetic pathway. J Bacteriol 170(10):4431–4436
Steinbuchel A, Schlegel HG (1991) Physiology and molecular genetics of poly(beta-hydroxy-alkanoic acid) synthesis in Alcaligenes eutrophus. Mol Microbiol 5(3):535–542
Steinbuchel A, Hustede E, Liebergesell M, Pieper U, Timm A, Valentin H (1992) Molecular basis for biosynthesis and accumulation of polyhydroxyalkanoic acids in bacteria. FEMS Microbiol Rev 9(2–4):217–230
Steinbuchel A, Fuchtenbusch B (1998) Bacterial and other biological systems for polyester production. Trends Biotechnol 16(10):419–427
Tsai PS, Nageli M, Bailey JE (2002) Intracellular expression of Vitreoscilla hemoglobin modifies microaerobic Escherichia coli metabolism through elevated concentration and specific activity of cytochrome o. Biotechnol Bioeng 79(5):558–567
Wang Q, Yu H, Xia Y, Kang Z, Qi Q (2009) Complete PHB mobilization in Escherichia coli enhances the stress tolerance: a potential biotechnological application. Microb Cell Fact 8:47
Wang Q, Liu X, Qi Q (2014) Biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) from glucose with elevated 3-hydroxyvalerate fraction via combined citramalate and threonine pathway in Escherichia coli. Appl Microbiol Biotechnol 98(9):3923–3931
Xie L, Hall D, Eiteman MA, Altman E (2003) Optimization of recombinant aminolevulinate synthase production in Escherichia coli using factorial design. Appl Microbiol Biotechnol 63(3):267–273
Yang JE, Choi YJ, Lee SJ, Kang KH, Lee H, Oh YH, Lee SH, Park SJ, Lee SY (2014) Metabolic engineering of Escherichia coli for biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) from glucose. Appl Microbiol Biotechnol 98(1):95–104
Yang P, Liu W, Cheng X, Wang J, Wang Q, Qi Q (2016) A new strategy for production of 5-aminolevulinic acid in recombinant Corynebacterium glutamicum with high yield. Appl Environ Microbiol 82(9):2709–2717
Yu X, Jin H, Cheng X, Wang Q, Qi Q (2016) Transcriptomic analysis for elucidating the physiological effects of 5-aminolevulinic acid accumulation on Corynebacterium glutamicum. Microbiol Res 192:292–299
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31670047 and 31370085).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Rights and permissions
About this article

Cite this article
Zhang, X., Zhang, J., Xu, J. et al. Engineering Escherichia coli for efficient coproduction of polyhydroxyalkanoates and 5-aminolevulinic acid. J Ind Microbiol Biotechnol 45, 43–51 (2018). https://doi.org/10.1007/s10295-017-1990-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-017-1990-4