
Abstract
Isoamyl alcohol can be used not only as a biofuel, but also as a precursor for various chemicals. Saccharomyces cerevisiae inherently produces a small amount of isoamyl alcohol via the leucine degradation pathway, but the yield is very low. In the current study, several strategies were devised to overproduce isoamyl alcohol in budding yeast. The engineered yeast cells with the cytosolic isoamyl alcohol biosynthetic pathway produced significantly higher amounts of isobutanol over isoamyl alcohol, suggesting that the majority of the metabolic flux was diverted to the isobutanol biosynthesis due to the broad substrate specificity of Ehrlich pathway enzymes. To channel the key intermediate 2-ketosiovalerate (KIV) towards α-IPM biosynthesis, we introduced an artificial protein scaffold to pull dihydroxyacid dehydratase and α-IPM synthase into the close proximity, and the resulting strain yielded more than twofold improvement of isoamyl alcohol. The best isoamyl alcohol producer yielded 522.76 ± 38.88 mg/L isoamyl alcohol, together with 540.30 ± 48.26 mg/L isobutanol and 82.56 ± 8.22 mg/L 2-methyl-1-butanol. To our best knowledge, our work represents the first study to bypass the native compartmentalized α-IPM biosynthesis pathway for the isoamyl alcohol overproduction in budding yeast. More importantly, artificial protein scaffold based on the feature of quaternary structure of enzymes would be useful in improving the catalytic efficiency and the product specificity of other enzymatic reactions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Due to global energy and environmental concerns, microbial production of biofuels from renewable resources has gained tremendous interest in recent years. Ethanol, the most conventionally used biofuel, is not an ideal fuel, because it has a lower energy density than gasoline and its high hygroscopicity raises the problem for storage and distribution [5, 30]. Higher alcohols, such as isobutanol and isoamyl alcohol, on the other hand, have higher energy densities, are not hygroscopic, and are less volatile compared to ethanol. Currently, the majority of the work on C4 and C5 alcohol production is carried out in prokaryotic hosts, such as Escherichia coli and Clostridium strain [4, 5, 10, 14, 15, 20, 21, 38]. Given that the yeast fermentation is relatively inexpensive and the downstream processing is well developed, Saccharomyces cerevisiae can be a suitable host for the industrial-scale production of higher alcohols. Unfortunately, there are only a few reports on engineering yeast S. cerevisiae for higher alcohol productions and the reported product yields are much lower compared to that attained in E. coli [6, 9, 24, 29, 37]. For example, the best isobutanol yield obtained in budding yeast was approximately 16 mg per g glucose, whereas it was reported to reach 162 mg per g glucose in E. coli [7, 9].
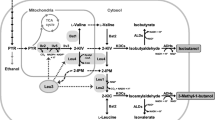
Isoamyl alcohol can be used not only as a biofuel, but also as a precursor for various chemicals, such as isoamyl acetate. S. cerevisiae naturally produces small amounts of isoamyl alcohol and other branched-chain alcohols as by-products during the fermentation [16, 19]. Recent works have focused on identifying gene targets, mutations, and pathways for increasing isoamyl alcohol production [1, 34]. As a by-product during the fermentation process, isoamyl alcohol is synthesized through the breakdown of leucine via the Ehrlich degradation pathway [19]. As shown in Fig. 1, isoamyl alcohol can also be produced directly from pyruvate in a biosynthetic pathway that recruits the upstream pathway of leucine biosynthesis. The upstream pathway from pyruvate to α-ketoisocaproate (KIC) comprises of acetolactate synthase (encoded by ILV2), ketol-acid reductoisomerase (encoded by ILV5), dihydroxyacid dehydratase (encoded by ILV3), 2-isopropylmalate (α-IPM) synthase (encoded by LEU4 or LEU9), isopropylmalate isomerase (encoded by LEU1), and 2-isopropylmalate dehydrogenase (encoded by LEU2). The downstream pathway from KIC to isoamyl alcohol comprises α-ketoacid decarboxylase (encoded by ARO10) and alcohol dehydrogenase (encoded by ADH7).

Schematic diagram of a cytosol-based orthogonal α-IPM biosynthetic pathway for isoamyl alcohol overproduction in S. cerevisiae. ILV2, encoding the mitochondrial acetolactate synthase; ILV5, encoding the mitochondrial acetohydroxyacid reductoisomerase; ILV3, encoding the mitochondrial dihydroxyacid dehydratase; LEU4 and LEU9, encoding α-IPM synthase; LEU1, encoding isopropylmalate isomerase; LEU2, encoding β-IPM dehydrogenase; ARO10, encoding ketoacid decarboxylase; ADH7, encoding alcohol dehydrogenase; MPC1-2/3, encoding the mitochondrial pyruvate carrier; ILV1, encoding threonine deaminase; OAC1, encoding the mitochondrial α-IPM transporter; BAT1, encoding the mitochondrial branched-chain amino-acid aminotransferase; BAT2, encoding the cytosolic branched-chain amino-acid aminotransferase. ILV2c, ILV5c, ILV3c, and LEU9c indicate genes with removal of the mitochondrial targeting signal sequence. The pathway intermediates α-IPM, β-IPM, and KIC are defined as 2-isopropylmalate, 3-isopropylmalate, and α-ketoisocarpoate, respectively. The dotted line indicates the translocation of metabolites between mitochondria and the cytosol. The blue arrow indicates the cytosolic α-IPM biosynthetic pathway. The green arrow indicates isoamyl alcohol synthesis from α-IPM. The red arrow indicates the synthesis of by-product of isobutanol and 2-methyl-1-butanol
Unlike prokaryotes, the native isoamyl alcohol biosynthetic pathway in budding yeast is complicated by subcellular compartmentalization. The upstream part of pathway from pyruvate to α-IPM is confined to mitochondria, whereas the downstream part of pathway from α-IPM to KIC and the Ehrlich degradation pathway are present in the cytosol [19]. In previous studies, isoamyl alcohol producing strain was constructed in its natural compartments by overexpressing some of the leucine biosynthetic pathway genes to increase isoamyl alcohol production [29]. However, simple overexpression of leucine biosynthetic pathway genes in their natural compartments to improve the isoamyl alcohol titer is limited by the translocation of intermediates between mitochondria and the cytosol. To achieve isoamyl alcohol overproduction in budding yeast, two main strategies are adopted in the current study: 1. Rewire a cytosol-based orthogonal α-IPM biosynthetic pathway to avoid the metabolite translocation between mitochondria and the cytosol and 2. Cytosolic expression of the downstream Ehrlich degradation pathway to minimize the formation of by-product (2-methyl-1-butanol). In addition, we also sought to develop an artificial protein scaffold to pull dihydroxyacid dehydratase and α-IPM synthase into a close proximity to channel the key intermediate KIV towards α-IPM biosynthesis and to further reduce the formation of by-product isobutanol.
Materials and methods
Strains and reagents
Escherichia coli strain DH5α was used for general plasmid constructions, and the strain was cultivated at 37 °C in Luria–Bertani medium with 100 μg/mL ampicillin. S. cerevisiae BY4741 was used as the parental strain for all yeast strain constructions. This strain was cultured in rich YPD medium. Engineered strains with different auxotrophic selection markers were grown in synthetic complete (SC) medium with different dropouts. For induction of genes under the control of galactose-inducible promoters, S. cerevisiae strains were grown in 3.6% galactose plus 0.4% glucose. Restriction enzymes, Taq polymerase, alkaline phosphatase (CIP), and T4 ligase were purchased from New England Biolabs (Beverly, MA). iProof HF polymerase and iScript™ Reverse Transcription Supermix were obtained from BioRad (Hercules, CA). Gel extraction kit, PCR purification kit, Plasmid purification kit, and RNeasy Mini Kit were purchased from QIAGEN (Hilden, Germany). FastStart Essential DNA Green Master Mix was purchased from Roche (Singapore, SG). All chemicals used in this study were purchased from Sigma-Aldrich (St. Louis, MO).
Plasmid construction
Oligonucleotides used for plasmid construction are listed in Table 1. The previously developed δ-integration platform [39] was used to construct a chromosome-based orthogonal (Table 2) leucine biosynthetic pathway under galactose-inducible promoters (GAL1/10 promoter). To create the genome integration cassette, a series of plasmids was constructed as follows. First, ILV2c, ILV5c, ILV3c, LEU9c, LEU1, and LEU2 were PCR amplified from the genomic DNA of S. cerevisiae BY4741. The internal BamHI restriction site in LEU2 gene was removed using overlap extension PCR. All PCR products were digested using BamHI/XhoI, and inserted into plasmid pδBLE2.0, to yield plasmid pδBLE2.0-ILV2c/ILV5c, pδBLE2.0-ILV3c/LEU9c, and pδBLE2.0-LEU2/LEU1. Next, these plasmids were used as template for the PCR amplification of genome integration cassettes using the primer pair Delta_IntF and Delta_IntR and the purified PCR product was transformed into BY4741 according to the previous study [39].
For the plasmid-based expression of the Ehrlich degradation pathway, ARO10 and ADH7 genes were PCR amplified from the genomic DNA of S. cerevisiae BY4741. The PCR product of ARO10 gene was digested with BamHI/XhoI, and inserted into pESC-URA derivative with a modified version of multiple cloning sites, to yield pRS426-ARO10. Next, the PCR product of ADH7 gene was digested with BglII/XhoI, and inserted into pRS426-ARO10 cut with BamHI/XhoI, to yield plasmid pRS426-ARO10/ADH7.
For the plasmid-based expression of protein scaffold, the ILV3c and LEU9c genes were PCR amplified from the genomic DNA of S. cerevisiae BY4741. The PCR product of ILV3c was digested with BamHI/HindIII, and the PCR product of LEU9c was digested with HindIII/XhoI. Both fragments were ligated into plasmid pSH62 [18] cut with BamHI/XhoI. The resulting plasmid with ILV3c and LEU9c in frame fused together was designated as pRS413-Scaffold1. The plasmid pRS413-Scaffold2 with the fusion of LEU9c and ILV3c was constructed in a similar way.
PCR verification of genome integration events
For the verification of genome integration events, colonies were first picked from the library and streaked on phleomycin-containing plates to eliminate false positives. Cells were lysed by 20 mM NaOH for 30 min in 100 °C water bath. PCR program was set as follows: 1 cycle of 95 °C for 5 min; amplification, 30 cycles of 95 °C for 15 s, 50 °C for 30 s, and 68 °C for 90 s; and 1 cycle of 68 °C for 3 min. Universal primer pair of F_pGAL1Scr and R_tCYC1Scr was used to verify the genome integration events of ILV5c, LEU9c, and LEU1. Universal primer pair of F_pGAL10Scr and R_tADH1Scr was used for the PCR verification of ILV2c, ILV3c, and LEU2. Only colonies with three-band pattern with the correct size corresponding to each gene were considered to have the orthogonal cytosolic leucine biosynthetic pathway assembled into yeast chromosomes.
RNA extraction and quantitative real-time PCR
Yeast cells were harvested at the early log phase, and a total amount of 1 × 107 cells was used for the total RNA extraction using the RNeasy Mini Kit (QIAGEN, Germany). Approximately 500 ng of RNA was converted to cDNA using iScript™ Reverse Transcription Supermix from Biorad (Hercules, CA).
Gene-specific primers for IVL2, ILV5, ILV3, LEU9, LEU1, LEU2, and ACT1 were designed using the ProbeFinder (https://www.roche-applied-science.com), and oligonucleotides used for qRT-PCR experiment are listed in Table 1. Quantitative PCR analysis was carried out using LightCycler 96 real-time machine with FastStart Essential DNA Green Master Mix (Roche) according to the manufacturer’s instructions. Each 20 μL reaction contained 50 ng of total cDNA, 10 μL FastStart Essential DNA Green Master Mix, and 0.5 μM of each primer. Thermal cycling conditions were set as follows: pre-incubation, 1 cycle of 95 °C for 10 min; amplification, 45 cycles of 95 °C for 10 s, 57 °C for 10 s, and 72 °C for 10 s. ACT1 was chosen as a reference housekeeping gene, and results were presented as ratios of gene expression between IVL2, ILV5, ILV3, LEU9, LEU1, and LEU2 and the reference gene, ACT1 [31].
Isoamyl alcohol production in engineered yeast cells
Strains harboring pRS426-ARO10/ADH7 were inoculated into SC-URA medium supplemented with 2% glucose. The next day, 14 mL tubes containing 2 mL SC-URA medium (3.6% galactose + 0.4% glucose) were inoculated with fresh cell cultures to an initial OD600 of 0.05. All tubes were immediately added with 20% (vol/vol) dodecane after seeding, to perform two-phase fermentation and harvest isoamyl alcohol. To investigate the effect of protein scaffold on isoamyl alcohol production, strains harboring pRS426-ARO10/ADH7 were further transformed with pRS413-Scaffold1 or pRS413-Scaffold2. Experiments were carried out in SC-URA-HIS medium (3.6% galactose + 0.4% glucose). The isoamyl alcohol level was measured after 72 h cultivation. Each time, 100 μL of cell culture was taken for measuring OD600 by microplate reader (Synergy H1, BioTek, USA), and 50 μL dodecane layer was sampled and diluted in 450 μL dodecane for the identification of isoamyl alcohol using gas chromatography-mass spectrometry (GC–MS). For the GC–MS analysis, 1 μL of diluted sample was injected into Shimadzu QP2010Ultra system equipped with a DB-5 ms column (30 m × 250 μm × 0.25 μm thickness) (Agilent Technologies, USA). Split ratio was set with 15:1. Helium (ultra purity) was used as carrier gas at a flow rate 1.0 mL/min. GC oven temperature was initially held at 40 °C for 2 min, ramped with a gradient of 5 °C/min until 45 °C and held for 4 min. The temperature was then raised with a gradient 15 °C/min until 230 °C and held for 4 min. Both the injector and detector were maintained at 225 °C. Scan mode was used to detect the mass range 40–120 m/z.
Results
Rewiring the cytosolic α-IPM synthesis pathway for production of isoamyl alcohol in budding yeast
In budding yeast, the leucine biosynthetic pathway is complicated by subcellular compartmentalization: the upstream part of pathway from pyruvate to α-IPM is confined into mitochondria, whereas the downstream part of pathway from α-IPM to KIC is confined in the cytosol (Fig. 1). The starting substrate of pyruvate is generated in the cytosol via the glycolysis and then transported into mitochondria by the mitochondrial pyruvate carrier (encoded by MPC1-2/3). As can be seen from Fig. 1, pyruvate not only serves as the substrate for the biosynthesis of valine and leucine, but also for the biosynthesis of isoleucine. More importantly, valine, leucine, and isoleucine biosynthetic pathways share the same enzymatic steps catalyzed by ILV2, ILV5, and ILV3. Overexpression of these enzymatic steps (ILV2, ILV5, and ILV3) in their native compartment would not only lead to a higher level of isoamyl alcohol production, but also result in an increased level of the unwanted by-product of 2-methyl-1-butanol. Therefore, rewiring the α-IPM synthesis pathway in the cytosol is expected to reduce the formation of 2-methyl-1-butanol. Moreover, this strategy also eliminates the translocation of metabolites between mitochondria and the cytosol, which is expected to further boost the pathway catalytic efficiency.
For this purpose, the N-terminal mitochondrial targeting sequence of ILV2, ILV5, and ILV3 is removed to achieve the cytosolic expression of these enzymes to catalyze the biosynthesis of KIV in the cytosol [9]. S. cerevisiae has two paralogous genes encoding α-IPM synthase, namely, LEU4 and LEU9. Previous biochemical studies revealed that LEU4 is a leucine-sensitive isoform, while LEU9 is resistant to such feedback inhibition [25]. Therefore, LEU9 is chosen for reconstructing the cytosol-based α-IPM biosynthetic pathway in budding yeast and the N-terminal 30 amino acids containing the mitochondrial targeting sequence was removed to enable the cytosol-relocalized LEU9. To further improve the isoamyl alcohol production, we also sought to overexpress the downstream part of the leucine biosynthetic pathway, namely, LEU1 and LEU2. The entire cytosolic leucine biosynthetic pathway was assembled into yeast chromosomes using the δ-integration platform developed in the previous study [39].
As shown in Fig. 2, six randomly isolated variants showed significantly high amounts of isobutanol, with the isobutanol product titer ranging from 400 to 750 mg/L. In contrast, only 200 ~ 300 mg/L of isoamyl alcohol was produced, indicating that the majority of KIV intermediate was diverted to isobutanol by Ehrlich degradation enzymes due to their broad substrate specificity. In comparison, the reference strain BY4741 with a plasmid-based expression of ARO10 and ADH7 only produced less than 20 mg/L of branched-chain alcohols (data not shown). Notably, only a small amount of 2-methyl-1-butanol was produced in our engineered yeast cells, and the level was significantly lower compared to isobutanol and isoamyl alcohol. These findings clearly indicated that the cytosol-relocalized α-IPM pathway is effective in reducing the formation of by-product 2-methyl-1-butanol. In the present study, the best branched-chain alcohol producer CC6a yielded approximately 275.83 ± 12.12 mg/L isoamyl alcohol, along with 743.38 ± 32.02 mg/L isobutanol and 88.20 ± 4.86 mg/L 2-methyl-butanol.

Isoamyl alcohol production in engineered yeast cells with a cytosol-based orthogonal α-IPM biosynthetic pathway. Strains CC1 ~ CC6 were transformed with plasmid pRS426-ARO10/ADH7, to yield CC1a–CC6a. All strains were cultivated in 14 mL tubes supplemented with 2 mL SC medium with additional valine, leucine, isoleucine, and uracil dropped out. The alcohol production in the engineered strains was measured after 72 h of cultivation. Data represented the average and standard deviation of three independent experiments
Further quantitative real-time PCR (qRT-PCR) analysis of a representative strain (strain CC6a) revealed that all leucine biosynthetic pathway genes were highly expressed when compared to that of the reference strain (Fig. 3). Notably, there were different expression levels for all the leucine biosynthetic pathway genes, suggesting that the pathway was combinatorially assembled into yeast chromosomes using the antibiotic-assisted δ-integration platform [39, 42]. In the future, strains with an even better performance could be isolated from the library, and further qRT-PCR analysis of the gene expression profile to identify the optimal copy number in these variants would shed some light on the rational design of pathway.

Quantitative real-time PCR analysis of the gene expression profile in the engineered yeast cells. The reference strain BY4741 and a representative strain CC6a were subjected to qRT-PCR analysis. Fresh overnight cultures were inoculated in SC media at an initial OD600 of 0.05 and harvested during the early log phase for qRT-PCR analysis. The results are presented as the relative abundance of ILV2, ILV5, ILV3, LEU9, LEU1, and LEU2 in each strain with respect to that of ACT1. Data represented the average and standard deviation of three independent experiments
Artificial protein scaffold for improving the isoamyl alcohol production
Based on the observation that there was a significant amount of isobutanol by-product formed by different strains, we next proceeded to develop an artificial protein scaffold to channel the key intermediate KIV towards α-IPM synthesis and further reduce the formation of by-product isobutanol. Traditionally, protein fusion is a widely used strategy to pull enzyme active sites into the close proximity and to minimize the diffusion of intermediates to the bulk phase [33, 35]. However, due to the complicated quaternary structure of metabolic enzymes, simple fusion of two enzymes may not only disturb the formation of the proper quaternary structure, but also lead to protein aggregates (Fig. 4a).

Effect of protein scaffold on isoamyl alcohol production in the engineered yeast cells. a Schematic diagram of fusion protein as an artificial protein scaffold to pull the connected reactions into the close proximity. Conventional protein fusion technology (Left); Fusion protein as a scaffold for recruiting free enzymes (Right). S, substrate; I, intermediates; P, product. b Alcohol production by different strains with the scaffold ILV3c-LEU9c. c Alcohol production by different strains with the scaffold LEU9c-ILV3c. All strains were cultivated in 14 mL tubes supplemented with 2 mL SC medium with additional histidine, valine, leucine, isoleucine, and uracil dropped out. The alcohol production in the engineered strains was measured after 72 h of cultivation. Data represented the average and standard deviation of three independent experiments
In this study, we attempted to utilize the feature of quaternary structure of metabolic enzymes to explore the fusion protein as a scaffold (Fig. 4a). By simply introducing a fusion protein into an engineered strain with individual enzyme overexpressed under GAL1/10 promoters, the fusion protein would serve as a scaffold to recruit free enzymes to form a multienzyme complex. Theoretically, the expression level of scaffold would affect the system performance in a bell-shape curve, as excess amounts of scaffold would also result in protein aggregates and have a deleterious effect on the catalytic efficiency of the connected reactions. The optimal ratio between the scaffold and free enzymes can be achieved by modulating the expression level of individual enzymes using the antibiotic-assisted multiple integration technique [39, 40].
As shown in Fig. 4b, expression of the ILV3c-LEU9c fusion protein in our engineered yeast cells resulted in an obvious change of the alcohol distribution and majority of these variants improved the isoamyl alcohol production by an additional twofold. Interestingly, both isobutanol and isoamyl alcohol in strains CC1b, CC4b, and CC5b were increased after expressing the scaffold, indicating that an additional overexpression of the scaffold ILV3c-LEU9c has a synergistic effect on the isobutanol production. In comparison, there was a decrease in the isobutanol level in strains CC2b, CC3b, and CC6b suggesting that the scaffold has a better impact on diverting KIV towards the isoamyl alcohol production. Among these variants, the best producer CC1b yielded approximately 472.24 ± 40.82 mg/L isoamyl alcohol, together with around 622.87 ± 28.12 mg/L isobutanol and 85.49 ± 24.36 mg/L 2-methyl-1-butanol (Fig. 4b). Notably, the majority of these variants have an increased level of the total amount of branched-chain alcohols, whereas 2-methyl-1-butanol level remained nearly unchanged.
In addition, we also investigated the fusion of ILV3c and LEU9c in an opposite orientation and the resulting scaffold was designated as LEU9c-ILV3c. As shown in Fig. 4c, the reverse orientation of scaffold could also improve the isoamyl alcohol production and minimize the formation of isobutanol by-product. More importantly, scaffold LEU9c-ILV3c improved isoamyl alcohol to a much higher level in majority of these variants, indicating that scaffold LEU9c-ILV3c might form a more favorable multienzyme complex and bring the enzyme active sites to an even closer proximity. The best producer CC3c produced 522.76 ± 38.88 mg/L isoamyl alcohol, together with 540.30 ± 48.26 mg/L isobutanol and 82.56 ± 8.22 mg/L 2-methyl-1-butanol.
Discussions
Despite the enormous success achieved in the E. coli research, branched-chain alcohol production in S. cerevisiae is still falling behind achievements made in the bacterial system [2, 8, 9, 22, 28]. Current efforts on the metabolic engineering of S. cerevisiae still heavily focus on overexpressing native pathway genes, balancing redox cofactors, and knocking-out competing pathways to improve the production of branched-chain alcohols [13, 26, 29]. Native isoamyl alcohol pathway has complicated subcellular compartmentalization [6], which leads to the issue of metabolite translocation and results in a low yield of isoamyl alcohol. To circumvent this issue, we rewired a cytosol-based orthogonal α-IPM biosynthetic pathway to avoid the metabolite translocation between mitochondria and the cytosol. To do this, the mitochondrial targeting signal sequence in ILV2, ILV5, ILV3, and LEU9 is removed to enable the cytosol-relocalized expression of these enzymes. Moreover, the downstream part of the leucine biosynthetic pathway that contains LEU1 LEU2 together with the Ehrlich degradation pathway (α-ketoacid decarboxylase encoded by ARO10, and alcohol dehydrogenase encoded by ADH7) were overexpressed to increase the pathway activity towards the isoamyl alcohol production.
As shown from Fig. 2, we found that all engineered yeast cells with a cytosol-based orthogonal α-IPM biosynthetic pathway coupled with the Ehrlich degradation pathway produced significantly higher amounts of isoamyl alcohol over the reference strain. However, due to the substrate promiscuity of downstream Ehrlich degradation enzymes, there was a significant amount of isobutanol co-formed during the fermentation process. Previous investigations revealed that high amounts of 2-methyl-1-butanol were produced in yeast cells when ILV2, ILV5, and ILV3 together with ARO10/ADH7 were overexpressed [6]. In contrast, there was only a small amount of 2-methyl-1-butanol formed in our engineered yeast cells, indicating that the cytosol-based leucine biosynthetic pathway is effective to reduce the formation of by-product 2-methyl-1-butanol.
In the present study, we also developed an artificial protein scaffold by employing the feature of quaternary structure of metabolic enzymes. Protein scaffolds have been previously reported to improve the catalytic efficiency of rate-limiting steps and increase the product formation [17, 27]. Co-localization of enzymes on RNA scaffolds also showed promising results on increasing the metabolite production in a geometrically dependent manner [32]. By pulling the enzyme active sites into the close proximity, KIV was successfully channeled towards α-IPM synthesis and the isoamyl alcohol level was improved by more than twofold in majority of these variants. Moreover, we found that additional expression of fused metabolic enzymes also had a synergistic effect on the overall pathway activity, as the total amount of branched-chain alcohols was increased in majority of variants. When compared to the conventional protein scaffold strategy [17, 27], our approach offers the advantage for an easy implementation, as it does not require the fusion of individual metabolic enzyme to other interaction domains. Nevertheless, the artificial protein scaffold strategy could be further applied by fusion of proteins-of-interest to other self-associated proteins to form an even more complex multienzyme complex.
In the present study, the best isoamyl alcohol producer yielded 522.76 ± 38.88 mg/L isoamyl alcohol, together with 540.30 ± 48.26 mg/L isobutanol and 82.56 ± 8.22 mg/L 2-methyl-1-butanol. However, the yield is still below commercial levels, and much lower compared to that attained in E. coli [15]. As we observed that there were still high amounts of ethanol formed during the fermentation process, blocking the ethanol formation would allow more metabolic flux towards the isoamyl alcohol production. Ideally, isoamyl alcohol could be served as the alternative redox sink over ethanol, which might open a new avenue to achieve the theoretical yield. To enable an industrial-level production of isoamyl alcohol, strategies, such as fine-tuning pathway components [3, 11, 12], deleting competing pathways [23], and developing metabolic valves and dynamic control devices [36, 41], might be employed to improve the strain performance. In the future, we will also focus on designing a ketoacid decarboxylase with better substrate specificity towards KIC to further reduce the formation of by-product isobutanol. Previously, cofactor engineering has been successfully established in E. coli, where the isobutanol production reached nearly 100% of the theoretical yield under anaerobic conditions [7]. As acetohydroxyacid reductoisomerase encoded by ILV5 in the upstream part of pathway exclusively uses NADPH, incorporation of cofactor regeneration system is expected to further improve the isoamyl alcohol titer by avoiding imbalanced levels of NADH/NADPH.
Conclusions
In summary, we established a novel strategy for the isoamyl alcohol overproduction in S. cerevisiae. Our work represents the first study to bypass the native compartmentalized α-IPM biosynthesis pathway and eliminate the metabolite transportation between mitochondria and the cytosol to boost the pathway activity. More importantly, the strategy of artificial protein scaffold would also find a broad utility for improving the catalytic efficiency of other connected enzymatic reactions.
Abbreviations
- α-IPM:
-
2-isopropylmalate
- β-IPM:
-
3-isopropylmalate
- KIC:
-
α-ketoisocarpoate
- KIV:
-
α-ketoisovalerate
- KMV:
-
2-keto-3-methyl-valerate
- KDC:
-
α-ketoacid decarboxylase
- ADH:
-
Alcohol dehydrogenase
- qRT-PCR:
-
Quantitative real-time PCR
- GC–MS:
-
Gas chromatography-mass spectrometry
References
Abe F, Horikoshi K (2005) Enhanced production of isoamyl alcohol and isoamyl acetate by ubiquitination-deficient Saccharomyces cerevisiae mutants. Cell Mol Biol Lett 10:383–388
Ai H, Shaner N, Cheng Z, Tsien R, Campbell R (2007) Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochem 46:5904–5910
Ajikumar PK, Xiao WH, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G (2010) Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science 330:70–74. doi:10.1126/science.1191652
Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJ, Hanai T, Liao JC (2008) Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng 10:305–311. doi:10.1016/j.ymben.2007.08.003
Atsumi S, Hanai T, Liao JC (2008) Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451:86–89. doi:10.1038/nature06450
Avalos JL, Fink GR, Stephanopoulos G (2013) Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat Biotechnol 31:335–341. doi:10.1038/nbt.2509
Bastian S, Liu X, Meyerowitz JT, Snow CD, Chen MMY, Arnold FH (2011) Engineered ketol-acid reductoisomerase and alcohol dehydrogenase enable anaerobic 2-methylpropan-1-ol production at theoretical yield in Escherichia coli. Metab Eng 13:345–352. doi:10.1016/J.Ymben.2011.02.004
Bermejo LL, Welker NE, Papoutsakis ET (1998) Expression of Clostridium acetobutylicum ATCC 824 genes in Escherichia coli for acetone production and acetate detoxification. Appl Environ Microbiol 64:1079–1085
Brat D, Weber C, Lorenzen W, Bode HB, Boles E (2012) Cytosolic re-localization and optimization of valine synthesis and catabolism enables inseased isobutanol production with the yeast Saccharomyces cerevisiae. Biotechnol Biofuels 5:65. doi:10.1186/1754-6834-5-65
Cann AF, Liao JC (2008) Production of 2-methyl-1-butanol in engineered Escherichia coli. Appl Microbiol Biotechnol 81:89–98. doi:10.1007/s00253-008-1631-y
Cao YX, Xiao WH, Liu D, Zhang JL, Ding MZ, Yuan YJ (2015) Biosynthesis of odd-chain fatty alcohols in Escherichia coli. Metab Eng 29:113–123. doi:10.1016/j.ymben.2015.03.005
Cao YX, Xiao WH, Zhang JL, Xie ZX, Ding MZ, Yuan YJ (2016) Heterologous biosynthesis and manipulation of alkanes in Escherichia coli. Metab Eng 38:19–28. doi:10.1016/j.ymben.2016.06.002
Chen X, Nielsen KF, Borodina I, Kielland-Brandt MC, Karhumaa K (2011) Increased isobutanol production in Saccharomyces cerevisiae by overexpression of genes in valine metabolism. Biotechnol Biofuels 4:21. doi:10.1186/1754-6834-4-21
Connor MR, Cann AF, Liao JC (2010) 3-Methyl-1-butanol production in Escherichia coli: random mutagenesis and two-phase fermentation. Appl Microbiol Biotechnol 86:1155–1164. doi:10.1007/s00253-009-2401-1
Connor MR, Liao JC (2008) Engineering of an Escherichia coli strain for the production of 3-methyl-1-butanol. Appl Environ Microbiol 74:5769–5775. doi:10.1128/AEM.00468-08
Dickinson JR, Harrison SJ, Hewlins MJ (1998) An investigation of the metabolism of valine to isobutyl alcohol in Saccharomyces cerevisiae. J Biol Chem 273:25751–25756
Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KL, Keasling JD (2009) Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol 27:753–759. doi:10.1038/nbt.1557
Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30:e23
Hazelwood LA, Daran JM, van Maris AJ, Pronk JT, Dickinson JR (2008) The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Appl Environ Microbiol 74:2259–2266. doi:10.1128/AEM.02625-07
Higashide W, Li Y, Yang Y, Liao JC (2011) Metabolic engineering of Clostridium cellulolyticum for production of isobutanol from cellulose. Appl Environ Microbiol 77:2727–2733. doi:10.1128/AEM.02454-10
Huo YX, Cho KM, Rivera JG, Monte E, Shen CR, Yan Y, Liao JC (2011) Conversion of proteins into biofuels by engineering nitrogen flux. Nat Biotechnol 29:346–351. doi:10.1038/nbt.1789
Inui M, Suda M, Kimura S, Yasuda K, Suzuki H, Toda H, Yamamoto S, Okino S, Suzuki N, Yukawa H (2008) Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli. Appl Microbiol Biotechnol 77:1305–1316. doi:10.1007/s00253-007-1257-5
Kondo T, Tezuka H, Ishii J, Matsuda F, Ogino C, Kondo A (2012) Genetic engineering to enhance the Ehrlich pathway and alter carbon flux for increased isobutanol production from glucose by Saccharomyces cerevisiae. J Biotechnol 159:32–37. doi:10.1016/J.Jbiotec.2012.01.022
Lee WH, Seo SO, Bae YH, Nan H, Jin YS, Seo JH (2012) Isobutanol production in engineered Saccharomyces cerevisiae by overexpression of 2-ketoisovalerate decarboxylase and valine biosynthetic enzymes. Bioproc Biosyst Eng 35:1467–1475. doi:10.1007/S00449-012-0736-Y
Lopez G, Quezada H, Duhne M, Gonzalez J, Lezama M, El-Hafidi M, Colon M, Martinez de la Escalera X, Flores-Villegas MC, Scazzocchio C, DeLuna A, Gonzalez A (2015) Diversification of paralogous alpha-isopropylmalate synthases by modulation of feedback control and hetero-oligomerization in Saccharomyces cerevisiae. Eukaryot Cell 14:564–577. doi:10.1128/EC.00033-15
Matsuda F, Ishii J, Kondo T, Ida K, Tezuka H, Kondo A (2013) Increased isobutanol production in Saccharomyces cerevisiae by eliminating competing pathways and resolving cofactor imbalance. Microb Cell Fact 12:119. doi:10.1186/1475-2859-12-119
Moon TS, Dueber JE, Shiue E, Prather KL (2010) Use of modular, synthetic scaffolds for improved production of glucaric acid in engineered E. coli. Metab Eng 12:298–305. doi:10.1016/j.ymben.2010.01.003
Nielsen DR, Leonard E, Yoon SH, Tseng HC, Yuan C, Prather KL (2009) Engineering alternative butanol production platforms in heterologous bacteria. Metab Eng 11:262–273. doi:10.1016/j.ymben.2009.05.003
Park SH, Kim S, Hahn JS (2014) Metabolic engineering of Saccharomyces cerevisiae for the production of isobutanol and 3-methyl-1-butanol. Appl Microbiol Biotechnol 98:9139–9147. doi:10.1007/s00253-014-6081-0
Peralta-Yahya PP, Keasling JD (2010) Advanced biofuel production in microbes. Biotechnol J 5:147–162. doi:10.1002/biot.200900220
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Sachdeva G, Garg A, Godding D, Way JC, Silver PA (2014) In vivo co-localization of enzymes on RNA scaffolds increases metabolic production in a geometrically dependent manner. Nucleic Acids Res 42:9493–9503. doi:10.1093/nar/gku617
Sarria S, Wong B, Martin HG, Keasling JD, Peralta-Yahya P (2014) Microbial synthesis of pinene. ACS synthetic biology 3:466–475. doi:10.1021/sb4001382
Schoondermark-Stolk SA, Jansen M, Veurink JH, Verkleij AJ, Verrips CT, Euverink GJ, Boonstra J, Dijkhuizen L (2006) Rapid identification of target genes for 3-methyl-1-butanol production in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 70:237–246. doi:10.1007/s00253-005-0070-2
Shiue E, Prather KL (2014) Improving D-glucaric acid production from myo-inositol in E. coli by increasing MIOX stability and myo-inositol transport. Metab Eng 22:22–31. doi:10.1016/j.ymben.2013.12.002
Solomon KV, Sanders TM, Prather KL (2012) A dynamic metabolite valve for the control of central carbon metabolism. Metab Eng 14:661–671. doi:10.1016/j.ymben.2012.08.006
Steen EJ, Chan R, Prasad N, Myers S, Petzold CJ, Redding A, Ouellet M, Keasling JD (2008) Metabolic engineering of Saccharomyces cerevisiae for the production of n-butanol. Microb Cell Fact 7:36. doi:10.1186/1475-2859-7-36
Yan Y, Liao JC (2009) Engineering metabolic systems for production of advanced fuels. J Ind Microbiol Biotechnol 36:471–479. doi:10.1007/s10295-009-0532-0
Yuan J, Ching CB (2015) Combinatorial assembly of large biochemical pathways into yeast chromosomes for improved production of value-added compounds. ACS Synth Biol 4:23–31. doi:10.1021/sb500079f
Yuan J, Ching CB (2014) Combinatorial engineering of mevalonate pathway for improved amorpha-4,11-diene production in budding yeast. Biotechnol Bioeng 111:608–617. doi:10.1002/bit.25123
Yuan J, Ching CB (2015) Dynamic control of ERG9 expression for improved amorpha-4,11-diene production in Saccharomyces cerevisiae. Microb Cell Fact 14:38. doi:10.1186/s12934-015-0220-x
Yuan J, Ching CB (2016) Mitochondrial acetyl-CoA utilization pathway for terpenoid productions. Metab Eng. doi:10.1016/j.ymben.2016.07.008
Acknowledgements
We would like to acknowledge the National University of Singapore for supporting the present study (NUS Start-up Grant: R279 000 364 133).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Yuan, J., Mishra, P. & Ching, C.B. Engineering the leucine biosynthetic pathway for isoamyl alcohol overproduction in Saccharomyces cerevisiae . J Ind Microbiol Biotechnol 44, 107–117 (2017). https://doi.org/10.1007/s10295-016-1855-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-016-1855-2