
Abstract
Muconic acid (MA) is a promising bulk chemical due to its extensive industrial applications in the production of adipic acid and other valuable, biodegradable intermediates. MA is heretofore mainly produced from petrochemicals by organic reactions which are not environmentally friendly or renewable. Biological production processes provide a promising alternative for MA production. We designed an artificial pathway in Escherichia coli for the biosynthesis of MA using the catechol group of 2,3-dihydroxybenzoate, an intermediate in the enterobactin biosynthesis pathway. This approach consists of two heterologous microbial enzymes, including 2,3-dihydroxybenzoate decarboxylase and catechol 1,2-dioxygenase. The metabolic flow of carbon into the heterologous pathway was optimized by increasing the flux from chorismate through the enterobactin biosynthesis pathway and by regulating the shikimate pathway. Metabolic optimization enabled a concentration of 605.18 mg/L of MA from glucose in a shaking flask culture, a value nearly 484-fold higher than that of the initial recombinant strain. The results indicated that the production of MA from this pathway has the potential for further improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Muconic acid (MA), also known as 2,4-hexadienedioic acid, has attracted great attention in recent years because of its potential use as a precursor and platform chemical for the production of several bio-plastics [5]. The products, including the commercially important bulk chemicals adipic acid, terephthalic acid, and trimellitic acid, have a wide variety of uses in the production of nylon-6, 6, polytrimethylene terephthalate, polyethylene terephthalate, dimethyl terephthalate, trimellitic anhydride, industrial plastics, pharmaceuticals, plasticizers, cosmetics, etc. [30]. Worldwide production of adipic acid has already exceeded 2.5 million tons per year [26]. Traditional chemical processes for MA production rely on non-renewable, petroleum-based feedstock and high concentrations of heavy metal catalysts, which results in environmental pollution, petroleum depletion, and high-cost separation processes [30]. Thus, a sustainable, environmentally friendly, and cost-effective biotechnological process for MA production, based on inexpensive carbohydrate raw materials, is very desirable.
MA is a naturally occurring metabolic product of some organisms, such as Acinetobacter sp., Pseudomonas sp., and Sphingobacterium sp. [17, 25, 28], which have metabolic pathways for the degradation of aromatic compounds. However, no known organisms can naturally produce MA from renewable carbon sources, such as glucose.
So far, four pathways have been designed for MA synthesis from glucose. Draths and Frost [6] reported the first synthetic route for MA production from glucose in Escherichia coli. This pathway was based on three heterologous genes, encoding 3-dehydroshikimate dehydratase (AroZ), protocatechuic acid decarboxylase (AroY) from Klebsiella pneumoniae, and catechol 1,2-dioxygenase (CatA) from Acinetobacter calcoaceticus, which convert 3-dehydroshikimate to MA via protocatechuic acid and catechol. Afterwards, the Frost group further optimized the strain by increasing the levels of phosphoenolpyruvate (PEP) and erythrose-4-phosphate (E4P), as well as by knocking out the 3-dehydroshikimate dehydrogenase gene aroE to block aromatic amino acid synthesis [18]. Recently, Apler’s and Boles’ groups reconstructed the same pathway to produce MA in Saccharomyces cerevisiae [5, 27]. Moreover, the Yan group assembled two more artificial pathways for MA production. In one of the pathways, tryptophan biosynthesis was shunted and MA was generated via anthranilate [23]. In the other, salicylic acid served as the precursor of MA, while salicylic acid was generated from isochorismate by isochorismate pyruvate lyase [13].
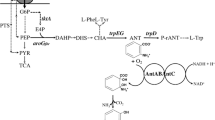
Some efforts have been made to study the mechanism of the enterobactin pathway [19, 22]. According to Franke and Bujnicki’s work, the fermentation of E. coli strains AN193/pDF3 (carrying entB and entC) and F111/pC20 (carrying aroF, entC, entB, aroB and aroL) showed that (S,S)-2,3-dihydroxy-2,3-dihydrobenzoic acid could be up to 4.6 and 12 g/L, respectively [4, 8], which indicates that the enterobactin pathway can also be practically applied to MA production. Very recently, the Yan group found a prokaryotic 2,3-dihydroxybenzoic acid decarboxylase and applied it to MA production [24]. In this study, we also successfully constructed a synthetic pathway to produce MA using the precursors of enterobactin, 2,3-dihydrobenzoate (2,3-DHB) (Fig. 1a). Two exogenous enzymes, 2,3-dihydroxybenzoate decarboxylase (EntX) and catechol 1, 2-dioxygenase (CatA) were imported from Klebsiella pneumonia strain CICIM B7001 and Pseudomonas putida strain KT2440, respectively. Metabolic optimization by increasing the flux enterobactin biosynthesis pathway and by regulating the shikimate pathway, as well as optimization of IPTG induction resulted in a concentration of 605.18 mg/L of MA from glucose in a shaking flask culture.

a. A metabolic pathway for the biosynthesis of MA. Light gray arrows indicate native metabolic pathways of chorismate in E. coli, dark gray arrows indicate practical metabolic pathways for enterobactin production in E. coli, and black arrows indicate the introduced artificial pathway for MA production. PEP phosphoenolpyruvic acid, E4P D-erythrose 4-phosphate, 2,3-DHB 2,3-dihydroxybenzoate, MA muconic acid, EntC isochorismate synthase, EntB isochorismatase; aroGfbr, encoding phenylalanine feedback inhibition-resistant 3-deoxy-d-arabinoheptulosonate 7-phosphate (DAHP) synthase, aroL encoding shikimate kinase, EntA 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase, EntX 2,3-dihydroxybenzoate decarboxylase, CatA catechol 1,2-dioxygenase. b. Gene organizations in the three cassettes. Genes entX, catA, entC, entBA, aroG fbr, and aroL encode EntX, CatA, EntC, EntBA, DAHP synthase, and shikimate kinase, respectively
Materials and methods
Microbial strains and plasmids
Escherichia coli strain JM109 was used for plasmid construction, while E. coli strain JM109 (DE3) was used for protein expression and MA production. Strains and primers used in this study are summarized in Tables 1 and 2. Standard cloning and bacterial transformations were performed according to Sambrook and Russell [20]. All restriction enzymes, DNA ligase, and the Prime STAR HS DNA Polymerase were purchased from TaKaRa (Dalian, China). The compatible vectors pETDuet-1, pCDFDuet-1, pRSFDuet-1, and pACYCDuet-1 were kindly provided by Prof. Zhou [29]. Plasmids used in this study are listed in Table 1. The gene entX (a hypothetical gene, named in this study, encoding 2,3-dihydroxybenzoate decarboxylase) was amplified from Klebsiella pneumonia CICIM B7001 genomic DNA by PCR using primers entX-F and entX-R. Additionally, the catA gene, encoding catechol 1,2-dioxygenase, was amplified from Pseudomonas putida KT2440 genomic DNA by PCR using primers catA-F and catA-R. Then, both entX and catA were sequentially cloned into vector pACYCDuet-1 using BglII and KpnI, and EcoRI and HindIII, respectively. The resulting plasmid was named pACYC-XA. pET-XA was constructed by digesting pACYC-XA and pETDuet-1 with EcoRI and KpnI, followed by ligation of the appropriate fragments.
The E. coli K-12 entC gene, encoding isochorismate synthase, was isolated by PCR using primers entC-F and entC-R. E. coli K-12 entBA, encoding isochorismatase and 2,3-dihydro-2, 3-dihydroxybenzoate dehydrogenase, was isolated by PCR using primers entBA-F and entBA-R. Then, both entC and entBA were sequentially cloned into vector pRSFDuet-1 using BglII and KpnI, and SacI and HindIII, respectively. The resulting plasmid was named pRSF-CBA.
E. coli K-12 aroG fbr, encoding a feedback inhibition-resistant 3-deoxy-d-arabinoheptulosonate 7-phosphate (DAHP) synthase, was isolated by fusion PCR using primers aroG-F, aroGm-down, aroGm-up, and aroG-R. E. coli K12 aroL, encoding shikimate kinase, was isolated by PCR using primers aroL-F and aroL-R. Then, both aroG fbr and aroL were sequentially cloned into vector pCDFDuet-1 using BglII and KpnI, and SacI and HindIII, respectively. The resulting plasmid was named pCDF-GL.
The plasmid pKD136, which expresses aroF (encoding 3-deoxy-7-phosphoheptulonate synthase), aroB (encoding 3-dehydroquinate synthase), and tktA (encoding transketolase), was isolated from strain E. coli ATCC 69875, which produces MA [6].
Strains Jm-1a and Jm-1b were constructed by transforming strain E. coli JM109 (DE3) with the plasmids pACYC-XA and pET-XA, respectively, to allow expression of entX and catA upon induction with isopropyl β-d-1-thiogalactopyranoside (IPTG). Similarly, strains Jm-2 and Jm-3 were constructed by transforming Jm-1a and Jm-1b with the plasmid pRSF-CBA, which expresses entC and entBA upon induction with IPTG. Strains Jm-4 and Jm-5 were constructed by transforming strain Jm-2 and Jm-3 with the plasmid pCDF-GL, which expresses aroG fbr and aroL upon induction with IPTG. Finally, strains Jm-6 and Jm-7 were created by transforming Jm-2 and Jm-4 with the plasmid pKD136, which expresses aroF, aroB, and aroL upon induction with IPTG.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis
Overnight culture of E. coli Jm-1a were inoculated into 30 mL LB medium and grew at 37 °C till OD660 reached 0.6, then induced with 0.20 mM IPTG. After 4 h induction, cells were harvested by centrifugation for 2 min at 12,000×g and suspended to 5 mL with 50 mM sodium phosphate buffer (pH 7.0), and treated by the ultrasonicator. SDS-PAGE was carried out on 12 % (w/v) polyacrylamide slab gels, which were stained with Coomasie brilliant blue R-250.
Reverse transcription-quantitative PCR (RT-qPCR)
Strains were grown in minimal medium. The cells were harvested by centrifugation for 2 min at 12,000×g and 4 °C and washed once with distilled water. The RNA was isolated using an RNAisoPlus kit (TaKaRa, Dalian, China). 1,000 ng of total RNA was treated with a iScript™ cDNA synthesis kit (Bio-Rad, Shanghai, China). The obtained cDNA was subjected to quantitative PCR (qPCR) using the LightCycler 2.0 system (Roche, Basel, Switzerland) and SYBR Premix Ex Taq (TaKaRa, Dalian, China). The 16S rRNA gene was set as reference for normalization. Primers used in RT-qPCR were listed in Table 2.
Culture conditions
LB medium (composed of 5 g/L yeast extract, 10 g/L peptone, and 10 g/L NaCl) and TB medium (composed of 24 g/L yeast extract, 12 g/L peptone, 40 g/L glycerol, 17 mM KH2PO4, and 72 mM K2HPO4) were used for inoculations and cell propagation. The synthetic minimal medium as described in [16] was used for MA production. Various combinations of ampicillin (100 mg/L), kanamycin (40 mg/L), chloramphenicol (20 mg/L), and streptomycin (40 mg/L) were added to cultures of plasmid-bearing E. coli strains. All strains were cultivated with 220 rpm orbital shaking. Strains were stored at −70 °C in 15 % glycerol.
Feeding experiments
Feeding experiments were conducted to examine the enzyme efficiency of EntX and CatA, which converted 2,3-DHB into MA. A single colony of strain Jm-1a, harboring pACYC-XA, was inoculated into 5 mL LB medium containing 20 μg/mL chloramphenicol and grown overnight at 37 °C. Two percent (v/v) of the overnight cultures were inoculated into 50 mL of TB medium containing 20 μg/mL chloramphenicol. The cultures were grown at 37 °C until the OD660 reached 0.6 and then they were induced at 30 °C with 0.20 mM IPTG. After 4 h, cells were harvested and re-suspended in 50 mL minimal medium. The cultures were supplemented with 2 mM 2,3-DHB and incubated at 30 °C with shaking, a 50 μL of 2 M 2,3-DHB solution was added into the cultures per hour. Samples were taken at 2, 4, 6, and 8 h. MA concentrations were analyzed by high-performance liquid chromatography (HPLC).
Microbial production of MA from glucose
Single colonies of the MA-producing strains were inoculated into 5 mL of LB medium containing appropriate antibiotics and grown overnight at 37 °C. Four percent (v/v) of the overnight cultures were inoculated into 50 mL minimal medium containing appropriate antibiotics and cultivated at 37 °C with shaking at 220 rpm in baffled shaker flasks. When the OD660 values of the cultures reached 0.6, IPTG was added to a final concentration of 0.20 mM to induce MA production. Samples were taken every 12 h. OD660 values were measured, and the products were analyzed by HPLC. All liquid cultivations were conducted in triplicate, at least.
HPLC analysis
Muconic acid (MA) (Sigma Aldrich, Shanghai, China) was used as the standard. Both the standard and samples were analyzed using a HPLC system (Waters, Milford, MA, USA) with a reverse-phase Agilent C18 column and a refractive index detector (Waters). A mobile phase of 0.33 mM H2SO4 solution at a flow rate 0.5 mL/min was applied to the column. The column temperature was set to 35 °C. Quantification of MA was based on the peak areas at 260 nm absorbance.
Results
Assembly of the MA biosynthetic pathway
In this study, a MA biosynthesis pathway using precursors of enterobactin was presented, which included the upstream pathway from glucose to shikimate to chorismate, the midstream pathway from chorismate to 2,3-DHB, and the downstream pathway from 2,3-DHB to MA (Fig. 1a). In this pathway, 2,3-DHB was regarded as the precursor of catechol that was subsequently transformed to MA, even though 2,3-DHB was the intermediate of enterobactin in E. coli, and chorismate was converted into 2,3-DHB by the enzymes EntC and EntBA. Considering there is no 2,3-DHB decarboxylase in E. coli, which converts 2,3-DHB into catechol, we assembled the downstream pathway of MA by co-expressing the heterologous genes entX and catA in E. coli. To accumulate 2,3-DHB from the chorismate pool, the partial enterobactin pathway (the midstream pathway) was reinforced by overexpressing entC and entBA. Moreover, to increase the carbon flow in the upstream pathway, a phenylalanine feedback inhibition-resistant gene, aroG fbr (which has a D146N mutation), encoding 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase, was cloned and co-expressed with shikimate kinase gene (aroL). The above genes, organized into three cassettes, are shown in Fig. 1b. Therefore, the MA biosynthetic pathway from glucose was constructed by relying upon the degradation of 2,3-DHB via EntX and CatA.
Bioconversion of 2,3-DHB to MA in E. coli
The downstream pathway from 2,3-DHB to MA was critical in the aforementioned MA biosynthetic pathway because E. coli lacks EntX activity, which converts 2,3-DHB into catechol. Earlier reports have shown that some fungi, such as Aspergillus niger and Trichosporon cutaneum, have EntX activity [2, 21]. Nevertheless, eukaryotic protein production in E. coli is limited because of problems associated with transcription, translation, and proper folding of proteins [7]. According to the KEGG database, the entX gene product of Klebsiella pneumoniae sub sp. pneumoniae NTUH-K2044 was defined as putative amidohydrolase, which was homologue 2,3-dihydroxybenzoate decarboxylase. A new literature of Yan group amplified entX from K. pneumonia, and verified the protein, which 35.9 % similarity with that from A. niger, showed decarboxylase activity [24]. In addition, catechol 1,2-dioxgenase from Pseudomonas putida strain KT2440 has a high CatA activity described by Sun et al. [13]. Therefore, to assemble the downstream pathway for MA production, we co-expressed the heterologous genes entX and catA in E. coli, the genes entX (999 bp) from K. pneumonia CICIM B7001 and catA (936 bp) from P. putida KT2440 were cloned into the double T7 expression vector pACYCDuet-1, resulting in the expression vector pACYC-XA (Fig. 2a), and thereby generating strain Jm-1a.

Identification of cloning and co-expressing EntX and CatA in E. coli. a Agarose gel electrophoresis of entX and catA amplified from pACYC-XA. Ma, 1 kb Ladder; Lane 1 entX gene of 999 bp, Lane 2 catA gene of 936 bp. b SDS-PAGE analysis of supernatant protein lysates. Mb, PageRuler Prestained Protein Ladder. Lane 1 recombinant EntX and CatA proteins, 35.7 and 36.1 kDa, respectively
SDS-PAGE analysis of supernatant protein lysates from strain Jm-1a showed that the two recombinant proteins (EntX and CatA) were expressed with molecular weight of 35.7 and 36.1 kDa, respectively (Fig. 2b), which were of the expected molecular weight similar with that reported by Sun et al. (EntX, 38 kDa band) [24] and Guzik et al. (CatA, 33.1 kDa band) [10]. Because 2,3-DHB was found toxic to the growth of E. coli cells in our previous work (see Supporting Information), a substrate feeding strategy was used to evaluate its potential in the bioconversion of 2,3-DHB to MA. Strain Jm-1a was pre-cultivated and transferred to the minimal medium containing 2,3-DHB. As seen in Fig. 3, by the end of 2, 4, 6, and 8 h incubation, the concentrations of MA reached 596.55, 1,173.92, 1,617.79, and 1,919.52 mg/L, respectively. The conversion rate of 2,3-DHB to MA reached about 100 % after 2 h and was 83.3 % after 8 h, while there is no MA detected by E. coli JM109 (DE3) harboring pACYCDuet-1, demonstrating the enzymatic efficiency of EntX and CatA with this plasmid. Accumulation of MA by reinforcing the enterobactin pathway and copy numbers of the XA cassette. Six genes, ent-A to -F, encode enzymes for the biosynthesis of enterobactin from the precursor chorismate. In the initial stage of enterobactin biosynthesis, the enzymes EntC, EntB, and EntA converted chorismate to 2,3-DHB. The remaining enzymes, Ent-D to -F, together with the bifunctional enzyme EntB, convert 2,3-DHB to enterobactin [15]. In E. coil, entC, entE, entB, and entA are adjacent on chromosome, according to the KEGG database. Besides, EntC is more active than its isozyme MenF [14]. Thus, we cloned entC, but not menF, and entBA into pRSFDuet-1, for the purpose of accumulating more 2,3-DHB.

Fed-batch experiment of the conversion of 2,3-DHB to MA by strain Jm-1a (filled square) and control E. coli JM109 (DE3) harboring pACYCDuet-1 (filled triangle). The data were generated from three independent experiments
As seen in Fig. 4, the titer of MA produced by strain Jm-1a, which only harbors pACYC-XA, was 1.25 mg/L. To increase flux through the enterobactin pathway from the chorismate pool, both EntC and EntBA were overexpressed by strain Jm-2, which resulted in a MA titer of 102.71 mg/L. Considering the aromatic compound 2,3-DHB, which is produced by EntCBA, is potentially toxic to E. coli [3], we further altered the copy number of the XA cassette by subcloning entX and catA from a low copy plasmid, pACYCDuet-1, to a relatively high copy number plasmid, pETDuet-1, thereby yielding strain Jm-3. As a result, MA production increased to 155.47 mg/L. The transcript levels of entX and catA in strain Jm-3 was much higher than those in Jm-2 (Fig. 5), which demonstrated that increase of copy numbers of XA cassette could improve the mRNA transcript levels of entX and catA, thereby, promote production of MA.

MA production from glucose by all recombinant strains used in this study. Strains are described in more detail in Table 1. All values were determined by HPLC after at least 72 h of batch flask cultures. Standard deviations are based on triplicate experiments

The transcription levels of entX and catA genes relative to 16S RNA in strains Jm-2 (gray) and Jm-3 (black). The 16S rRNA was set as 1.0. Standard deviations are based on triplicate experiments
Regulation of the shikimate pathway
In E. coli, the genes aroF, aroG, and aroH encode three 3-Deoxy-d-arabinoheptulosonate 7-phosphate (DAHP) synthase isozymes that are sensitive to tyrosine, phenylalanine, and tryptophan, respectively. Carbon flow through the shikimate pathway is regulated initially through repression and feedback inhibition by these three amino acids. In wild-type cells grown in minimal medium, Phe-, Tyr-, and Trp-sensitive DAHP synthases constitute about 80, 20, and 1 % of the total DAHP synthase activity, respectively [12]. Therefore, we overexpressed aroG fbr (which has a D146N mutation) in pCDFDuet-1 to relieve phenylalanine feedback repression. Additionally, shikimate kinase (aroL), which was reported to stimulate the shikimate pathway [4], was co-expressed in pCDFDuet-1 along with aroG fbr. Strains Jm-4 and Jm-5 were obtained, respectively. Furthermore, as the plasmid pKD136 from the strain ATCC 69875, which was reported to improve MA production [6], carries the genes encoding transketolase (tktA), DAHP synthase (aroF), and DHQ synthase (aroB), we also chose pKD136 as a candidate to regulate the shikimate pathway. We co-expressed pKD136 with the plasmids pRSF-CBA and pACYC-XA, thereby resulting in strain Jm-6.
Although pKD136 intensified the shikimate pathway, it failed to show a notable effect on MA accumulation in the presence of pACYC-XA and pRSF-CBA, which resulted in a MA yield by Jm-6 of only 111.75 mg/L. A possible reason for this low yield was that aroF in pKD136 does not relieve tyrosine feedback inhibition and relies upon weak, autologous promoters, and aroF comprised only about 20 % of the total DAHP synthase activity, while aroG comprised 80 % [12], which may has a much more poor effect on regulating shikimate pathway than pCDF-GL. Additionally, even when pCDF-GL was transformed into Jm-6, the titer of MA by Jm-7 was only 118.13 mg/L, which was not a significant improvement. The reason may be that pKD136 is a high copy number plasmid with a pBR322 origin, resulting in a metabolic burden with other 3 plasmids in Jm-7 from the expression of plasmid-borne genes and the replication of the plasmids. Strains Jm-4 and Jm-5, which overexpressed aroG fbr and aroL, showed a remarkable increase in MA production. Strain Jm-4 yielded up to 337.00 mg/L of MA, a 328 % increase compared to strain Jm-2, while the titer of MA in Jm-5 reached 456.77 mg/L, which was a 249 % increase by compared to Jm-3, and a 365-fold increase over Jm-1a (Fig. 4).
Relieving the metabolic burden of three plasmids in E. coli Jm-5
Because of expression of plasmid-borne resistance and replication of the plasmids, the three plasmids in E. coli Jm-5 could impose a metabolic burden on the host strain. One solution to alleviate the metabolic burden was to delay IPTG induction and change the concentration of IPTG [1, 11, 29]. Strain Jm-5 were cultured and induced by 0.20 mM IPTG at an OD660 value of 0.16, 0.63, 1.08, 1.66, 1.92, 2.30, or 3.02, respectively. As shown in Fig. 6, when IPTG induction was delayed, the final OD660 was increasing, but the production titer increased until an OD660 of 1.66. The addition of IPTG at an OD660 of 1.66 yielded the highest production titer (575.02 mg/L), and therefore, inductions in subsequent experiments were performed at an OD660 of 1.6.

Effect of IPTG induction at different cells growth stages (OD660 values) on MA production titers and final OD660 of strain Jm-5. MA (filled square), OD660 (filled column)
Based on the result of delaying IPTG induction, strains were induced at an OD660 value of 1.6 by different concentration of IPTG of 0, 0.10, 0.15, 0.20, 0.25, 0.30 or 0.40 mM. Figure 7 showed that low concentration of IPTG was beneficial to MA production. The highest production titer of MA reached 605.18 mg/L by 0.15 mM IPTG.

Effect of IPTG induction at different IPTG concentration on MA production titers and final OD660 of strain Jm-5. MA (filled square), OD660 (filled column)
Furthermore, the transcript levels of all expressed genes in the recombination strain Jm-5 were investigated. It can be seen from Fig. 8, comparing to control strain (E. coli JM109 (DE3) harboring pETDuet-1, pRSFDuet-1, and pCDFDuet-1) the transcript levels of overexpressed entC, entB, and entA in enterobactin biosynthesis pathway were increased more than 15 times, while that of aroG fbr and aroL in the shikimate pathway raised about five times. After optimization of IPTG induction, the transcript levels of aroG fbr and aroL, which related the regulating shikimate pathway improved about 50 %, and the genes encoding chorismate to MA were expressed more than 5–15 %, indicating the metabolic burden was lightened.

The transcription levels of aroG, aroL, entC, entB, entA, entX and catA relative to 16S RNA in strains control E. coli JM109 (DE3) harboring pETDuet-1, pRSFDuet-1, and pCDFDuet-1 (Light gray) and Jm-5. Jm-5 cells were induced at an OD660 value of 0.6 by 0.20 mM IPTG (dark gray), that of Jm-5* were at an OD660 value of 1.6 by 0.15 mM IPTG. The 16S rRNA was set as 1.0 (black). Standard deviations are based on triplicate experiments
Discussion
Muconic acid (MA) is a promising bulk chemical due to its extensive industrial applications [30]. In this study, we assembled a synthetic, composite pathway to produce MA by comprising two distinct enzymes, EntX and CatA. We also used several strategies to improve MA production. First, we strengthened the enterobactin pathway by overexpressing EntC and EntBA. Second, the copy numbers of the XA cassette were regulated. Then, the upstream pathway was intensified by overexpressing aroG fbr and aroL.
As can be observed, this pathway was based on the shikimate pathway, which leads to the biosynthesis of aromatic compounds that are essential in E. coli. Prior to this study, three artificial pathways derived from the shikimate pathway were reported for the microbial production of MA. Draths and Frost first reported the creation of metabolism of glucose to MA by introducing three heterologous enzymes, namely AroZ, AroY, and CatA, respectively, as well as disrupting shikimate dehydrogenase gene to accumulate more 3-dehydroshikimate. 16.8 mM of MA was produced in shake flask by E. coli AB2834/pKD136/pKD8.243A/pKD8.292 [6]. They further used this pathway to construct E. coil WN1/pWN2.248, which synthesized 36.8 g/L of MA after 48 h by culturing under fed-batch fermentations [18]. However, this blocked the shikimate pathway, which had the effect of turning the strain into an auxotroph for aromatic amino acids (phenylalanine, tyrosine, and tryptophan) and aromatic vitamins or vitamin-like intermediates (4-hydroxybenzoate, 4-aminobenzoate, and 2,3-dihydroxybenzoate) [6, 18]. Aromatic amino acids are relatively expensive, and their requirement would result in a large increase in the cost of MA production. Thus, there is also a need for a process that does not require these expensive nutrients to be added to the growth medium. Two studies used Saccharomyces cerevisiae to produce MA from glucose by the same pathway. However, the yields were 140 and 1.5 mg/L, respectively [5, 27]. In another MA producing pathway that shunted tryptophan biosynthesis from anthranilate, reported by the Yan group, there is a rate-limiting transamination step that converts chorismate into anthranilate. This reaction requires glutamine, which limited the efficiency of the whole pathway [23]. In the third pathway, MA was synthesized via salicylic acid. The yield of MA reached 1.5 g/L [13]. Although the formation of catechol from salicylic acid requires the coenzyme NADH.
Similarly to our work of MA production approach, the Yan group is the first to achieve the de nove production of 2,3-DHB and the conversion of 2,3-DHB to MA with plasmids pCS-entCBA-APTA and pZE-kpBDC-CDO, they assembled the two partial pathways in E. coil strain BW25113 ΔentE for microbial production of MA from glucose, and MA titer reached up to 480 mg/L at fermentation temperatures of 37 °C [24]. Here, we imported three cassette (pET-XA, pRSF-CBA, and pCDF-GL) to assemble strain Jm-5. Although the transcript levels of the expressed genes were not balanced (Fig. 5), the highest titer of MA reached 605.18 mg/L after optimization of IPTG induction. The transcriptional levels of the midstream pathway from chorismate to 2,3-DHB, and the downstream pathway from 2,3-DHB to MA was higher than the upstream pathway from glucose to shikimate to chorismate (Fig. 8), suggesting the upstream pathway may be continued to improve for production of MA.
Further work can also be performed on this pathway to enhance MA production. For example, AhpC, an alkyl hydroperoxide reductase, may participate in 2,3-DHB synthesis by increasing the availability of chorismate or increasing the efficiency with which EntC uses chorismate [15]. Thus, AhpC could be a potential key enzyme to improve MA production from this pathway. Gerstle et al. [9] reported that, RybA (one of the orphan sRNAs), downregulates aromatic amino acid biosynthesis under peroxide stress, and might increase the availability for other downstream products, such as products in the enterobactin pathway.
In summary, we constructed a MA biosynthetic approach by making use of the precursors of enterobactin. The final production of MA reached 605.18 mg/L, indicating that this biosynthetic pathway is promising for further modification and use in industrial MA production.
References
Alonso-Gutierrez J, Chan R, Batth TS, Adams PD, Keasling JD, Petzold CJ, Lee TS (2013) Metabolic engineering of Escherichia coli for limonene and perillyl alcohol production. Metab Eng 19:33–41
Anderson JJ, Dagley S (1981) Catabolism of tryptophan, anthranilate, and 2, 3-dihydroxybenzoate in Trichosporon cutaneum. J Bacteriol 146:291–297
Bartsch M, Bednarek P, Vivancos PD, Schneider B, von Roepenack-Lahaye E, Foyer CH, Kombrink E, Scheel D, Parker JE (2010) Accumulation of isochorismate-derived 2,3-dihydroxybenzoic 3-O-beta-d-xyloside in arabidopsis resistance to pathogens and ageing of leaves. J Biol Chem 285:25654–25665
Bujnicki RP, Bongaerts JJ, Raeven LJ, Sprenger G, Kuhm A, Takors R Improved biosynthetic production of 2, 3-trans-CHD. EP 1734130 A1
Curran KA, Leavitt JM, Karim AS, Alper HS (2013) Metabolic engineering of muconic acid production in Saccharomyces cerevisiae. Metab Eng 15:55–66
Draths KM, Frost JW (1994) Environmentally compatible synthesis of adipic acid from D-glucose. J Am Chem Soc 116:399–400
Finkelstein J, Antony E, Hingorani MM, O’Donnell M (2003) Overproduction and analysis of eukaryotic multiprotein complexes in Escherichia coli using a dual-vector strategy. Anal Biochem 319:78–87
Franke D, Lorbach V, Esser S, Dose C, Sprenger GA, Halfar M, Thömmes J, Müller R, Takors R, Müller M (2003) (S, S)-2,3-dihydroxy-2,3-dihydrobenzoic acid: microbial access with engineered cells of escherichia coli and application as starting material in natural-product synthesis. Chem Eeu J 9:4188–4196
Gerstle K, Klätschke K, Hahn U, Piganeau N (2012) The small RNA RybA regulates key-genes in the biosynthesis of aromatic amino acids under peroxide stress in E. coli. RNA Biol 9:458–468
Guzik U, Greń I, Hupert-Kocurek K, Wojcieszyńska D (2011) Catechol 1, 2-dioxygenase from rthe new aromatic compounds-degrading Pseudomonas putida strain N6. Int Biodeter Biodegr 65(3):504–512
Jones KL, Kim SW, Keasling JD (2000) Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metab Eng 2(4):328–338
Kikuchi Y, Tsujimoto K, Kurahashi O (1997) Mutational analysis of the feedback sites of phenylalanine-sensitive 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase of Escherichia coli. Appl Environ Microbiol 63:761–762
Lin Y, Sun X, Yuan Q, Yan Y (2014) Extending shikimate pathway for the production of muconic acid and its precursor salicylic acid in Escherichia coli. Metab Eng 23:62–69
Lin Y, Shen X, Yuan Q, Yan Y (2013) Microbial biosynthesis of the anticoagulant precursor 4-hydroxycoumarin. Nat Commun 4:2603. doi:10.1038/ncomms3603
Ma L, Payne SM (2012) AhpC is required for optimal production of enterobactin by Escherichia coli. J Bacteriol 194:6748–6757
Neidhardt FC, Bloch PL, Smith DF (1974) Culture medium for Enterobacteria. J Bacteriol 119:736–747
Neidle EL, Hartnett C, Bonitz S, Ornston LN (1988) DNA sequence of the Acinetobacter calcoaceticus catechol 1, 2-dioxygenase I structural gene catA: evidence for evolutionary divergence of intradiol dioxygenases by acquisition of DNA sequence repetitions. J Bacteriol 170:4874–4880
Niu W, Draths KM, Frost JW (2002) Benzene-free synthesis of adipic acid. Biotechnol Prog 18:201–211
Raymond KN, Dertz EA, Kim SS (2003) Enterobactin: an archetype for microbial iron transport. Proc Natl Acad Sci USA 100:3584–3588
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Santha R, Savithri HS, Rao NA, Vaidyanathan CS (1995) 2,3-Dihydroxybenzoic acid decarboxylase from Aspergillus niger: a novel decarboxylase. Eur J Biochem 230:104–110
Sikora AL, Wilson DJ, Aldrich CC, Blanchard JS (2011) Kinetic and inhibition studies of dihydroxybenzoate-AMP ligase (EntE) from Escherichia coli. Biochemistry 49:3648–3657
Sun X, Lin Y, Huang Q, Yuan Q, Yan Y (2013) A novel muconic acid biosynthesis approach by shunting tryptophan biosynthesis via anthranilate. Appl Environ Microbiol 79:4024–4030
Sun X, Lin Y, Yuan Q, Yan Y (2014) Biological production of muconic acid via a prokaryotic 2,3-dihydroxybenzoic acid decarboxylase. Chem Sus Chem. doi:10.1002/cssc.201402092
van Duuren JB, Wijte D, Leprince A, Karge B, Puchałka J, Wery J, Dos Santos VA, Eggink G, Mars AE (2011) Generation of a catR deficient mutant of P. putida KT2440 that produces cis, cis-muconate from benzoate at high rate and yield. J Biotechnol 156:163–172
van Duuren JB, Wittmann C (2014) Bioprocessing of renewable resources to commodity bioproducts. In: Bisaria VS, Kondo A (eds) First and second generation production of bio-adipic acid, 1st edn. Wiley, New York, pp 519–540
Weber C, Brückner C, Weinreb S, Lehr C, Essl C, Boles E (2012) Biosynthesis of cis, cis-muconic acid and its aromatic precursors, catechol and protocatechuic acid, from renewable feedstocks by Saccharomyces cerevisiae. Appl Environ Microbiol 78:8421–8430
Wu CM, Lee TH, Lee SN, Lee YA, Wu JY (2004) Microbial synthesis of cis, cis-muconic acid by Sphingobacterium sp. GCG generated from effluent of a styrene monomer (SM) production plant. Enzyme Microb Technol 35:598–604
Wu J, Du G, Zhou J, Chen J (2013) Metabolic engineering of Escherichia coli for (2S)-pinocembrin production from glucose by a modular metabolic strategy. Metab Eng 16:48–55
Xie NZ, Liang H, Huang RB, Xu P (2014) Biotechnological production of muconic acid: current status and future prospects. Biotechnol Adv 32:615–622
Acknowledgments
This work was supported by a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, the 111 Project (No. 111-2-06), and the Jiangsu province “Collaborative Innovation Center for Advanced Industrial Fermentation” industry development program.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, J., Zheng, P. Muconic acid production from glucose using enterobactin precursors in Escherichia coli . J Ind Microbiol Biotechnol 42, 701–709 (2015). https://doi.org/10.1007/s10295-014-1581-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1581-6