
Abstract
β-Mannanases (EC 3.2.1.78) can catalyze the cleavage of internal β-1,4-d-mannosidic linkages of mannan backbones, and they have found applications in food, feed, pulp and paper, oil, pharmaceutical and textile industries. Suitable amino acid substitution can promote access to the substrate-binding groove and maintain the substrate therein, which probably improves the substrate affinity and, thus, increases catalytic efficiency of the enzyme. In this study, to improve the substrate affinity of AuMan5A, a glycoside hydrolase (GH) family 5 β-mannanase from Aspergillus usamii, had its directed modification conducted by in silico design, and followed by site-directed mutagenesis. The mutant genes, Auman5A Y111F and Auman5A Y115F, were constructed by megaprimer PCR, respectively. Then, Auman5A and its mutant genes were expressed in Pichia pastoris GS115 successfully. The specific activities of purified recombinant β-mannanases (reAuMan5A, reAuMan5AY111F and reAuMan5AY115F) towards locust bean gum were 152.5, 199.6 and 218.9 U mg−1, respectively. The two mutants were found to be similar to reAuMan5A regarding temperature and pH characteristics. Nevertheless, the K m values of reAuMan5AY111F and reAuMan5AY115F, towards guar gum, decreased to 2.95 ± 0.22 and 2.39 ± 0.33 mg ml−1 from 4.49 ± 0.07 mg ml−1 of reAuMan5A, which would make reAuMan5AY111F and reAuMan5AY115F promising candidates for industrial processes. Structural analysis showed that the two mutants increased their affinity by decreasing the steric conflicts with those more complicated substrates. The results suggested that subtle conformational modification in the substrate-binding groove could substantially alter the substrate affinity of AuMan5A. This study laid a solid foundation for the directed modification of substrate affinities of β-mannanases and other enzymes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
β-Mannanases (endo-β-1,4-d-mannanases, EC 3.2.1.78) can cleave the internal β-1,4-d-mannosidic linkages of mannans, the major hemicellulosic constituents in cell walls of plants and some algae, and in some types of plant seeds [2]. They exist widely in bacteria, actinomycetes and fungi. Based on the analysis of primary structure and hydrophobic cluster, almost all β-mannanases have been classified into GH families 5, 26 and 113 (http://www.cazy.org/fam/acc_GH.html), which belong to GH clan-A. The overall three-dimensional (3-D) structure of clan-A β-mannanases consists principally of the (β/α)8 barrel fold [21].
β-Mannanases have drawn much attention owing to possessing great potential in various industrial applications, such as depolymerizing anti-nutritional factors in feeds, bleaching pulps, extracting oils, hydrolyzing mannan-based polymers used in hydraulic fracturing of oil and gas wells, and producing mannooligosaccharides [16]. Many studies have been performed on exploiting novel β-mannanases with good enzymatic properties, and on improving their activities by mutation breeding of enzyme-producing strains and optimization of expression conditions [17]. However, broad applicability of the majority of β-mannanases was limited by their weak substrate affinities, low activities or poor tolerance to extreme environments [9]. To meet the increasing demands for β-mannanases, more attention is being focused on modifying their primary and 3-D structures by genetic engineering. To date, the directed modifications of various glycoside hydrolases have been conducted to improve their substrate affinities, activities or thermostabilities, such as the Thermoascus aurantiacus endoglucanase [14], Aspergillus oryzae family 11 xylanase [4] and Thermoanaerobacter tengcongensis α-glucosidase [23]. Enhancing enzymatic substrate affinity can improve catalytic efficiency of the enzyme when the maximum reaction rate remains unchanged, and can make the catalytic reaction go more thoroughly. However, few papers have been reported about the directed modifications of β-mannanase substrate affinities or K m values.
Auman5A (GenBank accession No. HQ839639) encoding AuMan5A has been cloned from the genomic DNA of Aspergillus usamii YL-01-78 in our laboratory [15]. In this work, to improve the substrate affinity of AuMan5A, its directed modification was predicted by in silico design, including molecular docking simulation and binding free energy calculation. Based on this design, the mutant genes, Auman5A Y111F and Auman5A Y115F, were constructed by site-directed mutagenesis. Then, Auman5A and the mutant genes were expressed in Pichia pastoris GS115, respectively. Moreover, the enzymatic properties of reAuMan5A, reAuMan5AY111F and reAuMan5AY115F were characterized, especially in kinetic parameters. To our knowledge, this is the first report as to the directed modification of the A. usamii β-mannanase to improve its substrate affinity by in silico design and site-directed mutagenesis.
Materials and methods
Strains, vectors and culture media
Escherichia coli JM109 and pUCm-T vector (Sangon, Shanghai, China) were used for gene cloning. A recombinant T-vector, pUCm-T-Auman5A, was constructed and preserved in our lab [15]. The E. coli DH5α and pPIC9K vector (Invitrogen, USA) were used for construction of the recombinant expression vector. The E. coli JM109 and DH5α were cultured in the Luria–Bertani medium. P. pastoris GS115 and its transformants were cultured and induced in the YPD, MD, G418-containing YPD, BMGY and BMMY media, which were prepared as described in the manual of Multi-Copy Pichia Expression Kit (Invitrogen, USA).
Homology modeling of the β-mannanases and mannobiose
Based on the known crystal structure of a GH family 5 β-mannanase from Trichoderma reesei RutC-30 (PDB: 1QNR, 1.40 Å resolution), the 3-D structures of AuMan5A and its candidate mutants were homologically modeled using the MODELLER 9.9 program (http://salilab.org/modeller/). The 3-D structural information of mannobiose, which was used as the ligand, was handled using the GlycoBioChem PRODRG (http://davapc1.bioch.dundee.ac.uk/prodrg/submit.html).
Molecular docking simulation
The interaction of AuMan5A or its mutant with mannobiose was predicted by molecular docking (MD) simulation using the AutoDock 4.2 program (http://autodock.scripps.edu) to locate the most suitable binding position and orientation. Subsequently, the 3-D structures of molecule-docked complexes were optimized using the GROMACS 4.5 package (http://www.gromacs.org/). Based on the conformation of AuMan5A docking with mannobiose, all amino acids in the substrate-binding groove of AuMan5A and in proximity to mannobiose within 8 Å were located by PyMol software (http://pymol.org/).
Selection of the candidate mutant β-mannanases
Family 5 β-mannanases, which shared more than 50 % identities with AuMan5A, were searched in the NCBI website (http://blast.ncbi.nlm.nih.gov/) using the BLAST server. Then, the multiple sequence alignment of the searched β-mannanases (containing AuMan5A) was carried out using the ClustalW2 program (http://www.ebi.ac.uk/Tools/msa/clustalw2/). According to the physicochemical properties of non-conserved amino acids in proximity to mannobiose within 8 Å and their locations on the 3-D structure of AuMan5A, several amino acids were selected to be substituted by the similar ones or frequently emerging ones in other β-mannanases, forming a series of candidate mutant β-mannanases.
Calculation of the binding free energies
The binding free energy (ΔG bind) of a receptor (β-mannanase in this work) docking with a ligand (mannobiose), contrary to the affinity between them [6], was calculated using the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method [22]. The ΔG bind is defined as: ΔG bind = G complex − G receptor − G ligand, where the free energy (G) of a complex, receptor or ligand is calculated as: G = E MM − TSMM + G solv. The E MM is the molecular mechanics energy, which is mainly composed of the intramolecular electrostatic energy (E elec) and van der Waals energy (E vdW) and calculated by using the GROMACS 4.5 package. The entropy value (TSMM) can be neglected because the goal is to make comparisons of the ΔG bind between the mutants and AuMan5A [6]. The G solv is the solvation free energy, which consists principally of the polar energy (G PB) and non-polar solvation energy (G SUR) and is calculated by using the APBS package (http://www.poissonboltzmann.org/apbs/).
Construction of the mutant β-mannanase genes
The genes, Auman5A Y111F and Auman5A Y115F, were constructed by megaprimer PCR, respectively [18]. Four PCR primers were designed according to the nucleotide sequence of Auman5A and synthesized by Sangon (Shanghai, China) as listed in Table 1. The DNA fragment, YF111 or YF115, was first amplified from pUCm-T-Auman5A with primers AuMan5A-F and Y111F-R or Y115F-R by the following conditions: a denaturation at 94 °C for 3 min, 30 cycles at 94 °C for 30 s, 55 °C for 30 s, 72 °C for 25 s, and an elongation at 72 °C for 10 min. Using pUCm-T-Auman5A as the template again, the second-round PCR for the complete mutant gene, Auman5A Y111F or Auman5A Y115F, was carried out using YF111 or YF115 and AuMan5A-R as the primers under the same conditions, except for an elongation at 72 °C for 75 s in 30 cycles. The target PCR products were purified using the EZ-10 Spin Column DNA Gel Extraction Kit (BBI, Canada), and inserted into the pUCm-T vector, followed by transforming them into E. coli JM109, respectively. The recombinant T-vectors containing Auman5A Y111F and Auman5A Y115F, named pUCm-T-Auman5A Y111F and pUCm-T-Auman5A Y115F, were confirmed by DNA sequencing.
Transformation of the recombinant expression vectors
The three β-mannanase genes were excised from pUCm-T-Auman5A, pUCm-T-Auman5A Y111F and pUCm-T-Auman5A Y115F by digestion with EcoRI and NotI, and inserted into the pPIC9K vector digested with the same enzymes, followed by transforming them into E. coli DH5α, respectively. The recombinant vectors, named pPIC9K-Auman5A, pPIC9K-Auman5A Y111F and pPIC9K-Auman5A Y115F, were confirmed by DNA sequencing. Subsequently, the resulting recombinant vectors were separately linearized with SacI, and transformed into P. pastoris GS115 by electroporation using a Gene Pulser apparatus (Bio-Rad, USA) according to the manufacturer’s instruction.
Screening and expression of the Pichia pastoris transformants
All P. pastoris transformants were primarily screened based on their ability to grow on a MD plate, and successively inoculated on geneticin G418-containing YPD plates at increasing concentrations of 1.0, 2.0 and 4.0 mg ml−1 for the screening of multiple copies of integrated genes Auman5A, Auman5A Y111F and Auman5A Y115F, respectively. The P. pastoris GS115 transformed with pPIC9K vector was used as the negative control, numbered as P. pastoris GSC. Expression of the β-mannanase gene in P. pastoris GS115 was performed according to the instruction of Multi-Copy Pichia Expression Kit (Invitrogen, USA) with slight modification [9].
Purification of the expressed β-mannanases
After the P. pastoris transformant was induced by 1 % (v/v) methanol for 96 h, a total of 40 ml of cultured supernatant was brought to 75 % saturation by adding solid ammonium sulfate, and left overnight. The resulting precipitate was harvested, dissolved in 4 ml of 20 mM Na2HPO4–NaH2PO4 buffer (pH 6.0), and dialyzed against the same buffer overnight. The dialysate was concentrated to 1 ml by ultrafiltration using a 10-kDa cut-off membrane (Millipore, USA), and then loaded onto a Sephadex G-75 column (Amersham Pharmacia Biotech, Sweden; 1.6 × 80 cm), followed by elution with the same buffer at a flow rate of 0.3 ml min−1. Aliquots of 1.5 ml eluent only containing target reAuMan5A, reAuMan5AY111F or reAuMan5AY115F were pooled and concentrated for further studies.
Enzyme activity and protein assays
β-Mannanase activity was determined by incubating 0.1 ml of suitably diluted enzyme solution with 2.4 ml of 0.5 % (w/v) locust bean gum (LBG) (Sigma, USA) in 50 mM Na2HPO4–citric acid buffer (pH 3.6) at 55 °C for 10 min. The amount of released reducing sugars was measured with the 3,5-dinitrosalicylic acid (DNS) method [9], using d-mannose as the standard. One unit (U) of β-mannanase activity was defined as the amount of enzyme liberating 1 μmol of reducing sugar equivalent per minute under the standard assay conditions stated above. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out according to the method of Laemmli [7] on a 12.5 % gel. The isolated proteins were visualized by staining with Coomassie Brilliant Blue R-250 (Sigma, USA), and their apparent molecular weights were estimated using Quantity One software based on the standard protein markers. The protein concentration was measured with the BCA-200 Protein Assay Kit (Pierce, USA), using bovine serum albumin as the standard.
Enzymatic kinetic parameter assays
The hydrolytic reaction rate (U mg−1) of β-mannanase was assayed under the standard assay conditions, except the substrate (LBG, KGM: konjac glucomannan, guar gum) concentrations ranging from 1.0 to 10 mg ml−1. The reaction rate versus substrate concentration was plotted to confirm whether the hydrolytic mode of reAuMan5A, reAuMan5AY111F or reAuMan5AY115F conforms to the Michaelis–Menten equation. The K m and V max values were determined by non-linear regression analysis using the software Origin 8.
Results and discussion
In silico design of the mutant β-mannanases
Based on the docked conformation of AuMan5A and mannobiose, 38 amino acids of AuMan5A in proximity to mannobiose within 8 Å were located using the PyMol software. According to the results of Hogg et al. [5] and Sabini et al. [13], aromatic amino acids can form hydrophobic platforms, which are important for substrate binding. In this work, five aromatic amino acids, Trp54, Tyr111, Tyr115, Phe206 and Tyr243, were selected from 17 unconserved amino acid residues of AuMan5A, while Tyr216 was ignored for its low frequency of occurrence in other mannanases (Fig. 1). Then, each single aromatic amino acid was substituted by the corresponding one with a similar property or frequently emerging one in other β-mannanases, forming a series of candidate mutant β-mannanases. Six mutants together with AuMan5A are listed in Table 2. The MD simulation can rapidly find the most suitable binding position and orientation for a ligand on a given macromolecule (receptor). This methodology has been used as a powerful tool to predict the affinity between ligand and receptor [3], and to screen novel drugs such as human phospholipase A2 and HIV-1 protease inhibitors [12].

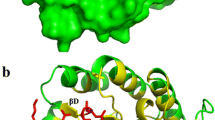
Detailed properties of unconserved amino acid residues of AuMan5A in proximity to mannobiose within 8 Å. The figure presents the results of the structural bioinformatics and evolutionary analysis. a Rows correspond to residue properties and columns correspond to specific positions. The individual residue properties are: the position index, amino acid type, secondary structure (H helix, S strand, C other), proximity to catalytic residues in Å, proximity to substrate in Å. The residue types and frequencies of the two most frequent amino acid residue types at each position are presented (— no other types). b Cartoon view of AuMan5A with mannobiose. c Surface view of AuMan5A with mannobiose
The ΔG bind values of AuMan5A and its candidate mutants docking with mannobiose were calculated by MM-PBSA, respectively. As a result, ΔG bind values of AuMan5AY111F and AuMan5AY115F docking with mannobiose were the lowest, which decreased to −56.8 and −75.0 kcal mol−1 from −47.7 kcal mol−1 of AuMan5A (Table 2). A distinct decrease in ΔG bind value may confer the high substrate affinity on AuMan5AY111F or AuMan5AY115F based on the analytical results by computational prediction as reported previously [6]. The site-directed mutagenesis of key amino acids based on the in silico design could modify enzymatic properties and/or activities purposefully, and avoid a substantial invalid or deadly mutation [24].
Construction of the mutant β-mannanase genes
To verify the prediction by in silico design, AuMan5A was separately mutated to AuMan5AY111F and AuMan5AY115F by megaprimer PCR. An about 350-bp specific band of fragment YF111 or YF115 was obtained by first-round PCR with primers AuMan5A-F and Y111F-R or Y115F-R (Fig. 2, lane 1 or 2). Then, the second-round PCR for a complete gene, Auman5A Y111F or Auman5A Y115F was performed with primers YF111 or YF115 and AuMan5A-R. As a result, two about 1,050-bp specific bands of mutant genes were amplified (Fig. 2, lanes 3 and 4), and inserted into the pUCm-T vector, respectively. The resulting recombinant T-vectors containing the correct inserts, designated pUCm-T-Auman5A Y111F and pUCm-T-Auman5A Y115F, were confirmed by DNA sequencing.

The mutant β-mannanase genes, Auman5A Y111F and Auman5A Y115F, constructed by megaprimer PCR. Lane M DNA marker, lane 1 or 2 the DNA fragment of YF111 or YF115 amplified by the first-round PCR, lane 3 or 4 the complete mutant gene of Auman5A Y111F or Auman5A Y115F amplified by the second-round PCR
Expression of the recombinant β-mannanases
The P. pastoris transformant that could resist the high concentration of geneticin G418 might have multiple copies of integration of a heterologous gene into P. pastoris genome, which could potentially lead to the high-level expression of a heterologous protein. However, the protein expression level was not directly proportional to the concentration of G418 [8]. Owing to those reasons, all P. pastoris transformants, containing three kinds of β-mannanase genes and resistant to 1.0, 2.0 and 4.0 mg ml−1 of G418, were picked out for flask expression tests. Among all transformants tested, three transformants expressing the maximum reAuMan5A, reAuMan5AY111F and reAuMan5AY115F activities of 44.8, 53.0 and 50.4 U ml−1, respectively, numbered as P. pastoris GSAuM4-8, GSAuM1114-11 and GSAuM1154-5, were screened. No β-mannanase activity was detected in the cultured supernatant of P. pastoris GSC under the same expression conditions.
Purification of the recombinant β-mannanases
One of advantages of the P. pastoris expression system was that the purity of expressed recombinant protein was very high as described in the manual of the Multi-Copy Pichia Expression Kit (Invitrogen, USA), which could greatly simplify the purification procedures. It was reported that purities of the recombinant A. usamii xylanase AuXyn11D and A. sulphureus β-mannanase expressed in P. pastoris GS115 and X-33 reached 90 and 97 %, respectively [1, 20]. In this work, the amount of reAuMan5A, reAuMan5AY111F or reAuMan5AY115F, secreted into the culture supernatant, accounted for more than 75 % of that of the total protein (Fig. 3, lanes 1, 2 or 3), which was assayed by protein band-scanning. Therefore, they were separately purified to homogeneity only by a simple combination of ammonium sulfate precipitation, ultrafiltration and a Sephadex G-75 gel chromatograph. SDS-PAGE analysis of the purified reAuMan5A, reAuMan5AY111F or reAuMan5AY115F displayed one single protein band with an apparent molecular weight of about 49.5 kDa (Fig. 3, lanes 4, 5 or 6). The specific activities of purified reAuMan5A, reAuMan5AY111F and reAuMan5AY115F, towards 0.5 % LBG under the standard assay conditions, were 152.5, 199.6 and 218.9 U mg−1, respectively. The specific activity of reAuMan5AY111F and reAuMan5AY115F separately increased by 30.9 % and 43.3 % compared with that of reAuMan5A.

SDS-PAGE analysis of reAuMan5A, reAuMan5AY111F and reAuMan5AY115F secreted by P. pastoris GSAuM4-8, GSAuM1114-11 and GSAuM1154-5, respectively. Lane M, protein marker; lane 1, 2 or 3, the cultured supernatant of GSAuM4-8, GSAuM1114-11 or GSAuM1154-5; lane 4, 5 or 6, the purified reAuMan5A, reAuMan5AY111F or reAuMan5AY115F with an apparent molecular weight of about 49.5 kDa
Temperature and pH characteristics of the recombinant β-mannanases
Thermoactivity and thermostability of the purified enzymes were determined and compared with those of the wild-type enzyme (data not shown). No distinct differences in the two aforementioned parameters were observed between the three enzymes. In fact, all enzymes showed maximum activities at 70 °C and retained over 85 % of their original activities at 55 °C or below when incubated at pH 3.6 and at varied temperatures for 1 h. Furthermore, no significant differences were observed in pH characteristics between all enzymes. Three recombinant β-mannanases showed the highest activities at pH 3.5 and were highly stable at a pH range of 3.0–7.0 when incubated at 40 °C and at various pH values for 1 h.
Kinetic parameters of the recombinant β-mannanases
The kinetic parameters of reAuMan5A, reAuMan5AY111F and reAuMan5AY115F towards LBG, KGM and guar gum were determined and are summarized in Table 3. The wild-type and the mutant enzymes had the highest affinity and catalytic efficiency toward LBG when compared with the other substrates, as did other β-mannanases [10, 19]. Nevertheless, K m values of reAuMan5AY111F and reAuMan5AY115F for guar gum decreased about 34 % and 47 %, the catalytic efficiencies increased 0.5-fold and 0.7-fold correspondingly as compared with those of reAuMan5A (Table 3). The binding affinity and catalytic efficiency of the mutant enzymes for KGM increased, but not as obviously as those for guar gum. These results showed that both Phe111 and Phe115 played an important role in substrate binding and catalysis.
Roles of mutations in the substrate binding
Guar gum has more side chains than LBG, and the content of β-d-galactopyranose residue in guar gum is about 33 %, much higher than that in LBG (20 %) [11], thus, the structure of guar gum is more complicated, which limits the access of it to the binding groove. Mutating amino acids near the catalytic sites with a large side chain to one with a small side chain may decrease the steric conflict with guar gum, which makes it easier to access and maintain guar gum in the catalytic cavity. Tyr111, proximity to the catalytic site is 3.1 Å (Fig. 1) and has the same orientation as mannobiose (Fig. 4), is an important amino acid for binding. Structure alignment demonstrated that Phe111 of AuMan5AY111F had a smaller side chain than Tyr111 of AuMan5A. Moreover, the amino acid polarity of this site changed, and a more hydrophobic residue may make this area more compact compared with the original form. The accessible molecular surface per residue was calculated by WHAT TF (http://swift.cmbi.ru.nl/servers/html/ index.html), and for this residue it decreased to 4.8 Å2 from 5.7 Å2, indicating that the decrease of K m after substitution was probably due to conferring side chains of complicated substrate more flexibility.

Superposition of residues Tyr111 from AuMan5A and Phe111 from AuMan5AY111F. The catalytic residues (Glu168 and Glu276), Tyr111 and Phe111 residues are shown as sticks, the mannobiose binds in subsites −1 and +1
Tyr115 was located at subsites −3 and −2, and formed a gate with Pro318. After mutation, the distance between Phe115 and Pro318 was 7.0 Å, while the distance between Tyr115 and Pro318 was 5.7 Å (Fig. 5), in other words, the gate was enlarged, which may make reAuMan5AY115F display lower K m values and higher catalytic efficiency towards those substrates with more complicated structures. These results demonstrated that subtle conformational modification in the substrate-binding groove could substantially decrease the K m value or increase the substrate affinity of AuMan5A, which were consistent with those reported by Hogg et al. [5] and Srikrishnan et al. [14].

Local model of the mutation sites and the catalytic residues in AuMan5A and AuMan5AY115F. Glu168, Tyr115 or Phe115, Glu276 and Pro318 residues are shown as sticks, the mannobiose binds in subsites −2 and −1. a A close-up view showing the distance from Tyr115 to Pro318 in AuMan5A. b A close-up view showing the distance from Phe115 to Pro318 in AuMan5AY115F
Conclusions
The mutant β-mannanase genes, Auman5A Y111F and Auman5A Y115F, were constructed by in silico design, and followed by site-directed mutagenesis. Subsequently, Auman5A, Auman5A Y111F and Auman5A Y115F were expressed in P. pastoris GS115, respectively. The site-directed mutagenesis of two key amino acids in the substrate-binding groove of AuMan5A obviously decreased its K m value for guar gum (from 4.49 ± 0.07 to 2.95 ± 0.22 and 2.39 ± 0.33 mg ml−1), having no significant alteration in temperature and pH characteristics. Structural analysis showed that the two mutations could increase the affinity of AuMan5A by decreasing the steric conflicts with those more complicated substrates. Our present work first established a novel strategy for molecular modification of the enzyme to improve its substrate affinity by in silico design.
References
Chen XL, Cao YH, Ding YH, Lu WQ, Li DF (2007) Cloning, functional expression and characterization of Aspergillus sulphureus β-mannanase in Pichia pastoris. J Biotechnol 128:452–461
Dhawan S, Kaur J (2007) Microbial mannanases: an overview of production and applications. Crit Rev Biotechnol 27:197–216
Dias R, Saraiva Macedo Timmers LF, Caceres RA, de Azevedo WF Jr (2008) Evaluation of molecular docking using polynomial empirical scoring functions. Curr Drug Targets 9:1062–1070
Gao SJ, Wang JQ, Wu MC, Zhang HM, Yin X, Li JF (2013) Engineering hyperthermostability into a mesophilic family 11 xylanase from Aspergillus oryzae by in silico design of N-terminus substitution. Biotechnol Bioeng 110:1028–1038
Hogg D, Woo EJ, Bolam DN, McKie VA, Gilbert HJ, Pickersgill RW (2001) Crystal structure of mannanase 26A from Pseudomonas cellulosa and analysis of residues involved in substrate binding. J Biol Chem 276:31186–31192
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33:889–897
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Li JF, Tang CD, Shi HL, Wu MC (2011) Cloning and optimized expression of a neutral endoglucanase gene (ncel5A) from Volvariella volvacea WX32 in Pichia pastoris. J Biosci Bioeng 111:537–540
Li JF, Zhao SG, Tang CD, Wang JQ, Wu MC (2012) Cloning and functional expression of an acidophilic β-mannanase gene (Anman5a) from Aspergillus niger LW-1 in Pichia pastoris. J Agric Food Chem 60:765–773
Lu H, Zhang H, Shi P, Luo H, Wang Y, Yang P, Yao B (2013) A family 5 β-mannanase from the thermophilic fungus Thielavia arenaria XZ7 with typical thermophilic enzyme features. Appl Microbiol Biotechnol 97:8121–8128
Moreira LRS, Filho EXF (2008) An overview of mannan structure and mannan-degrading enzyme systems. Appl Microbiol Biotechnol 79:165–178
Pérez C, Pastor M, Ortiz AR, Gago F (1998) Comparative binding energy analysis of HIV-1 protease inhibitors: incorporation of solvent effects and validation as a powerful tool in receptor-based drug design. J Med Chem 41:836–852
Sabini E, Schubert H, Murshudov G, Wilson KS, Siika-aho M, Penttilä M (2000) The three-dimensional structure of a Trichoderma reesei β-mannanase from glycoside hydrolase family 5. Acta Cryst D56:3–13
Srikrishnan S, Randall A, Baldi P, Da Silva NA (2012) Rationally selected single-site mutants of the Thermoascus aurantiacus endoglucanase increase hydrolytic activity on cellulosic substrates. Biotechnol Bioeng 109:1595–1599
Tang CD, Guo J, Wu MC, Zhao SG, Shi HL, Li JF, Zhang HM, Wang JQ (2011) Cloning and bioinformatics analysis of a novel acidophilic β-mannanase gene, Auman5A, from Aspergillus usamii YL-01-78. World J Microbiol Biotechnol 27:2921–2929
van Zyl WH, Rose SH, Trollope K, Görgens JF (2010) Fungal β-mannanases: mannan hydrolysis, heterologous production and biotechnological applications. Proc Biochem 45:1203–1213
Wu MC, Tang CD, Li JF, Zhang HM, Guo J (2011) Bimutation breeding of Aspergillus niger strain for enhancing β-mannanase production by solid-state fermentation. Carbohydr Res 346:2149–2155
Xie ZH, Shi XJ (2009) Fast and almost 100 % efficiency site-directed mutagenesis by the megaprimer PCR method. Prog Biochem Biophys 36:1490–1494
Xu M, Zhang R, Liu X, Shi J, Xu Z, Rao Z (2013) Improving the acidic stability of a β-mannanase from Bacillus subtilis by site-directed mutagenesis. Process Biochem 48:1166–1173
Zhang HM, Wu MC, Li JF, Gao SJ, Yang YJ (2012) Cloning and expression of a novel xylanase gene (Auxyn11D) from Aspergillus usamii E001 in Pichia pastoris. Appl Biochem Biotechnol 167:2198–2211
Zhang YL, Ju JS, Peng H, Gao F, Zhou C, Zeng Y, Xue YF, Li Y, Henrissat B, Gao GF, Ma YH (2008) Biochemical and structural characterization of the intracellular mannanase AaManA of Alicyclobacillus acidocaldarius reveals a novel glycoside hydrolase family belonging to clan GH-A. J Biol Chem 283:31551–31558
Zhang Y, Pan D, Shen Y, Jin N, Liu H, Yao X (2012) Understanding the molecular mechanism of the broad and potent neutralization of HIV-1 by antibody VCR01 from the perspective of molecular dynamics simulation and binding free energy calculations. J Mol Model 18:4517–4527
Zhou C, Xue YF, Ma YH (2010) Enhancing the thermostability of α-glucosidase from thermoanaerobacter tengcongensis MB4 by single proline substitution. J Biosci Bioeng 110:12–17
Zhu XL, Lai LH (2009) A novel method for enzyme design. J Comput Chem 30:256–267
Acknowledgments
This work was financially supported by the National Nature Science Foundation of China (No. 31271811), Doctoral Research Funds of Jiangnan University, China (No. JUDCF11011), and Postgraduate Innovation Training Project of Jiangsu, China (No. CXZZ11_0480). We are grateful to Prof. Xianzhang Wu (School of Biotechnology, Jiangnan University, Wuxi, Jiangsu, China) for providing technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, J., Wei, X., Tang, C. et al. Directed modification of the Aspergillus usamii β-mannanase to improve its substrate affinity by in silico design and site-directed mutagenesis. J Ind Microbiol Biotechnol 41, 693–700 (2014). https://doi.org/10.1007/s10295-014-1406-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1406-7