
Abstract
Microbes are the leading producers of useful natural products. Natural products from microbes and plants make excellent drugs. Significant portions of the microbial genomes are devoted to production of these useful secondary metabolites. A single microbe can make a number of secondary metabolites, as high as 50 compounds. The most useful products include antibiotics, anticancer agents, immunosuppressants, but products for many other applications, e.g., antivirals, anthelmintics, enzyme inhibitors, nutraceuticals, polymers, surfactants, bioherbicides, and vaccines have been commercialized. Unfortunately, due to the decrease in natural product discovery efforts, drug discovery has decreased in the past 20 years. The reasons include excessive costs for clinical trials, too short a window before the products become generics, difficulty in discovery of antibiotics against resistant organisms, and short treatment times by patients for products such as antibiotics. Despite these difficulties, technology to discover new drugs has advanced, e.g., combinatorial chemistry of natural product scaffolds, discoveries in biodiversity, genome mining, and systems biology. Of great help would be government extension of the time before products become generic.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Secondary metabolites from natural sources (microbes, plants, animals) have helped to double the human life span during the 20th century, combated pain and suffering, and revolutionized the practice of medicine [38]. Infectious disease was the leading cause of human death in the world in 1900 [125]. Recently, it has been the second-leading cause of death worldwide and number three in developed nations [75]. Microbes have provided compounds that have cured or reduced the effect of human diseases for 70 years. Microbial products include antibiotics against bacteria and fungi, antitumor drugs, immunosuppressants, enzyme inhibitors including hypocholesterolemics, antiparasitic agents, bioherbicides, plant growth regulators, biopesticides, and bioinsecticides. Other activities utilizing natural products that are in use or being studied include treatments for viral diseases, acne, malaria, prion diseases, diabetes, hyperlipoproteinemia, obesity, gastric ulcers, iron-overload disease (hemochromatosis) and alumina overload in kidney dialysis patients.
The value and occurrence of natural products
Microbes have been very important in the production of natural product drugs. Of 23,000 active compounds from microorganisms, i.e., antimicrobials, antivirals, cytotoxic, and immunosuppressive compounds, 42 % are made by fungi and 32 % by filamentous bacteria, the actinomycetes.
Ever since the discovery of penicillin by Alexander Fleming in 1928, its development in the early 1940s at Oxford by Chain, Florey, Heatley and Abraham, and the discovery of useful streptomycete products in the early 1940s at Rutgers University by Waksman, Woodruff, Schatz and Lechevalier, we have benefited from the remarkable selective action of antibiotics on pathogenic bacteria and fungi [39]. These include (but are not limited to) penicillins, cephalosporins, tetracyclines, aminoglycosides, chloramphenicol, macrolides, ansamycins, polyenes, and glycopeptides. Over half of the antibiotics are produced by the actinomycetes, 10–15 % by non-filamentous bacteria and about 20 % from filamentous fungi. Many useful products are semi-synthetic derivatives of the above compounds, which are produced by chemistry or by bioconversion.
Out of about 1 million natural products, approximately 25 % are biologically active, i.e., show positive activities or toxicity. About 60 % of these are from plants and most of the rest from microbes; some are from animal sources. We already know the structures of over 160,000 natural products, about half from plants and half from microbes. This figure grows at about 10,000 per year.
Although most known natural products have been obtained from terrestrial environments, 129 bioactive compounds were isolated from marine microbes from 2000 to 2003 [69]. It is clear that the marine environment has yielded many new natural products [58]. About 20,000 are known, despite the fact that <1 % of commensal microbiotic consortia of marine invertebrates are culturable. Bacteria occupy up to 40 % of the biomass of sponges [88]. Sponges are one of the most important sources of biologically active products, and the producers are often their bacterial symbionts. For example, the genes encoding the biosynthesis of antitumor polyketides known as onnamides and theopederins from the marine sponge Theonella swinhoei are prokaryotic in sequence. The genes are similar to those found in the terrestrial beetles which produce the antitumor polyketide pederin; here the production is due to an uncultured Pseudomonas symbiont. Thus, it appears that in both beetles and sponges, the polyketides are made by symbiotic bacteria.
Marine cyanobacteria produce many compounds with activities such as neurotoxic, antiproliferative, anticancer, and anti-infective. These are Gram-negative photoautotrophic prokaryotes carrying out oxygenic photosynthesis. They are known as blue-green bacteria due to their production of C-phycocyanin, a blue green pigment used for photosynthesis. Their products have anticancer, antibacterial, antiviral, immunomodulatory and protease-inhibition activities.
Approved marine products include Cytarabine (Cytostar®) for non-Hodgkin’s lymphoma, which was originally isolated from a sponge, Vidarabine (Vira-A®), Ziconotide (Prialt®) and Trabactedin (Yondelis®). Many of those in clinical trials are for cancer.
The first genome sequenced from an obligate marine actinobacterium was that of Salinispora tropica which makes salinosporamides, sporolides and lymphostatin. Genome sequencing revealed that 10 % of the genome was devoted to secondary metabolism [109]. At least 19 biosynthetic loci were detected that produced siderophores, polyketides, melanins, non-ribosomal and ribosomal peptides, terpenoids and aminocyclitols. Genome sequencing of another marine actinobacterium, Salinispora arenicola, revealed 39 different biosynthetic loci of natural products. Symbionts of marine invertebrate animals have also revealed interesting natural products [43]. Cyanovirin, a 101-amino-acid protein isolate from Nostoc ellipsosporium, is a fusion inhibitor of HIV and also inhibits influenza A and B viruses.
Natural products make excellent drugs. From 1981 to 2002, 60 % of new chemical entities (NCEs) for cancer were natural, as were 75 % of the NCEs for infectious disease. Natural products or compounds related to them constitute over 60 % of approved and pre-new drug application (NDAs) candidates, not including biologicals such as monoclonal antibodies or vaccines [76]. These include natural products (6 %), derivatives of natural products (27 %), synthetic compounds with natural product pharmacophores (5 %), and synthetic mimics of natural products (23 %). About 78 % of antibacterials and 74 % of antitumor agents are natural or related to natural products. Approximately half of the leading pharmaceutical products on the market are natural or related compounds. For example, from 1981 to 2002, natural products were the basis of 74 % of all new chemical entities for cancer, 48 out of 74 anti-hypertensive agents, and seven out of ten anti-migraine agents. Almost 50 % of new drugs introduced into the marketplace between 1985 and 2005 were natural or related products. In 2003, the market for such compounds amounted to $40 billion.
Certain structures are well represented among successful natural products, e.g., isoprenoids, alkaloids and polyketides. The number of known isoprenoids, including terpenoids and carotenoids, amount to about 50,000. The alkaloids number over 16,000 and the polyketides around 10,000. Most of the polyketides are produced by bacteria and fungi. Markets for such compounds included $12 billion for terpenoids [91] and $17 billion for polyketides.
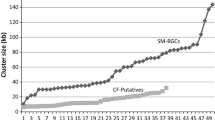
One microbe often produces many secondary metabolite compounds [13]. A gentamicin-producing strain of Micromonospora forms 50 isolatable secondary metabolites. Further bacterial examples include 12 compounds made by Streptomyces sp. Go40/14, 12 by sp. A1, 24 by sp Tn64, 30 by sp Go.40/10 and 32 by sp.Tn3634 [98]. A single strain of Myxococcus xanthus produces 38 different epothilones [59]. Aspergillus ochraceus produces 16 compounds. Another fungus, Sphaeropsideles sp F-24′707 produces 19 compounds. These high numbers were obtained by varying nutritional conditions, physical parameters or adding inhibitors. Many of the detected compounds were previously unknown, Biosynthetic genes of microorganisms are present in clusters coding for large multidomain and multimodular enzymes, e.g., polyketide synthases, prenyltransferases, non-ribosomal peptide synthases and terpene cyclases. By sequencing the Aspergillus nidulans genome, it was determined that the organism could produce 27 polyketides, 14 non-ribosomal peptides, one terpene, and two indole alkaloids [14]. Multiple gene clusters encoding secondary metabolites are common in species of Streptomyces, other filamentous actinomycetes and mycobacteria [23]. Streptomyces coelicolor and Streptomyces avermitilis contain 20–30 of these clusters. On the other hand, non-filamentous bacterial genomes seem to lack them. Genes adjacent to the biosynthetic gene clusters encode regulatory proteins, oxidases, hydroxylases and transporters. Strategies to activate silent genes have been recently reviewed [15].
Antibiotics
The selective action exerted on pathogenic bacteria and fungi by microbial secondary metabolites ushered in the antibiotic era and for over 50 years, we have benefited from this remarkable property of “wonder drugs”. Natural products have served us well in combating infectious bacteria and fungi. Microbial and plant secondary metabolites helped to double our life span during the 20th century, reduced pain and suffering, and revolutionized medicine. Most antibiotics are either (1) natural products of microorganisms, (2) semi-synthetically produced from natural products, or (3) chemically synthesized based on the structure of the natural products. Already known are 15,000 antibiotics with 150 on the market [111].
Antibiotics have been crucial in the increase in average life expectancy in the US from 47 years in 1900 to 74 for males and 80 for women in 2000 [67]. They have been virtually the only drugs utilized for chemotherapy against pathogenic microorganisms. They are defined as low molecular weight organic natural products (secondary metabolites or idiolites) made by microorganisms that are active at low concentration against other microorganisms. The most important microbiological antibiotics include the β-lactams (penicillins and cephalosporins), tetracyclines, aminoglycosides, chloramphenicol, macrolides, and glycopeptides.
The first medically useful antibiotic was discovered by Alexander Fleming in 1928 as a product of the fungus Penicillium notatum but its purification, isolation, and structure were not known for another 15 years. This was finally accomplished by the elegant work of Ernst Chain, Howard Florey, Norman Heatley, and Edward Abraham at Oxford University. In the early 1940s, the Selman Waksman group at Rutgers University, which included H. Boyd Woodruff, Albert Schatz and Hubert Lechevalier, discovered useful antibiotics produced by the actinomycetes, i.e., filamentous bacteria. These included actinomycin, the aminoglycosides (aminocyclitols) including streptomycin and neomycin, and many other antibiotics. After these successes, new compounds continued to be discovered. Benjamin Duggar’s group at Lederle Laboratories of American Cyanamid (now Pfizer) announced the discovery of chlortetracycline (aureomycin, biomycin) in 1948 as produced by Streptomyces aureofaciens. It was approved for use that year against both Gram-positive and Gram-negative bacteria. This was soon followed by the discovery at Pfizer of oxytetracycline (Terramycin). A completely different compound, chloramphenicol, was discovered by scientists at Parke-Davis (now Pfizer) in cooperation with workers at Yale University and University of Illinois. chloramphenicol is produced by Streptomyces venezuelae but has been made by chemical synthesis due to its relatively simple structure. Additional aminoglycosides were discovered such as kanamycin by Hamao Umezawa at the Institute for Microbial Chemistry in Tokyo and gentamicin by Marvin Weinstein and coworkers at Schering-Plough (now Merck) in New Jersey (USA). Aminoglycosides are broad-spectrum in activity, stable chemically and bactericidal.
For some unknown reason, the filamentous bacteria (actinomycetes) are amazingly prolific in the number of antibiotics they can produce [7]. About 75 % of the antibiotics are produced by actinomycetes and about 75 % of these are made by a single genus, Streptomyces. Strains of Streptomyces hygroscopicus make almost 200 antibiotics. One Micromonospora strain can produce 48 aminoglycoside antibiotics. Streptomyces griseus strains produce over 40 different antibiotics. Other organisms making antibiotics include strains of Bacillus subtilis producing over 60 such compounds. Twelve percent of the antibiotics are produced by non-filamentous bacteria and about 20 % are made by filamentous fungi. The antibiotics vary in size from small molecules like cycloserine (102 Daltons) and bacilysin (270 Daltons) to polypeptides, such as nisin which contains 34 amino acid residues. Myxobacterium xanthus devotes 9 % of its genome to production of secondary metabolites, which is twice that of S. coelicolor [44]. Myxobacteria, as a group, produce more than 300 antibiotics.
More than 350 agents have reached the world market as antimicrobials. They include (a) natural products, (b) semi-synthetic antibiotics and (c) synthetic chemicals [19]. The commercial antibiotics include the cephalosporins (45 %), penicillins (15 %), quinolones (11 %), tetracyclines (6 %), macrolides (5 %); the remainder include the aminoglycosides, ansamycins, glycopeptides, lipopeptides and polyenes. The strictly synthetics include the sulfa drugs, azoles, oxazolidinones (linezolid), quinolones, and fluoroquinolones.
There have been about 40 β-lactam compounds used in medicine. Despite the fact that β-lactamases are the major cause of resistance development and there are over 450 such enzymes, β-lactams are still very useful due to the discovery of β-lactamase inhibitors. These include clavulanic acid, and the carbapenems. The latter include doripenem (S-4661), which has broad spectrum activity including Pseudomonas aeruginosa, as well as tomopenem, ceftobiprole, ceftaroline, faropenem and meropenem.
Tuberculosis is about the worst disease caused by bacterial infection with Mycobacterium tuberculosis killing 1.6–2 million people each year. A combination of the carbapenem meropenem with clavulanic acid has been found to inhibit M. tuberculosis, even when the organism is in its “persistent” state, and also extensively drug-resistant strains [62]. Since they are both FDA-approved drugs, this combination is being used to treat patients with previously untreatable TB.
Tetracyclines have had a significant contribution to the antibiotic era. Chlortetracycline was discovered in 1948 followed by oxytetracycline in 1950. These were the first broad-spectrum antibiotics known. Resistance eventually developed via antibiotic efflux which was combated by the second-generation tetracyclines, i.e., minocycline and doxycycline, which were semi-synthetic, more lipophilic and thus taken up to a greater degree by the resistant strains, resulting in a decrease in net efflux. However, resistance based on ribosomal protection soon developed. These resistant strains were attacked by the third-generation tetracycline, the glycylcycline tigecycline, approved in 2005. Another third-generation tetracycline is PTIC-0796. Doxycycline hyclate (Periostat®) is also used for periodontal disease by inhibiting enzymes that breakdown gum tissue.
Polyketide macrolides are another group of useful antibiotics. The term macrolide is a shortened version of macrolactone glycoside which was proposed by the famous chemist R.B. Woodward in 1957. The term refers to a macrolactone containing one or more deoxy sugars. They are made by actinomycetes, e.g., Streptomyces, Micromonospora, Saccharopolyspora, and Actinoplanes. In the early 1950s, the polyketide macrolide erythromycin was approved for oral use in outpatients. It was later accompanied in medical use by other macrolides, i.e., oleandomycin, pikromycin, amphotericin B, midecamycin, josamycin, and carbomycin. Tylosin came along for use in animals. Their major problems were acid instability, poor bioavailability, and rapid elimination. Thus, they had to be given three or four times per day. Semi-synthetic second-generation macrolides followed, e.g., clarithromycin and azithromycin. Midecamycin was replaced by miokamycin and rokitamycin, while tylosin was improved upon by tilmicosin. Others included dirithromycin, florithromycin, and roxithromycin. These second-generation compounds were more acid-stable, eliminated from the body at a lower rate, and only required dosing at one or two times per day. Third-generation “ketolides” appeared in the late 1980s, mainly to overcome resistance to the earlier compounds. These included telithromycin, introduced in 2001 in Europe and in 2004 in the USA. Lipiarmycin (dificidin, tiacamycin B, OPT-80) is particularly active against Clostridium difficile infections and has been approved. Additional macrolides include spiramycin. The major targets of the macrolides are respiratory pathogens, sexually transmitted infections (Chlamidia trachomatis), Legionnaires’ disease (Legionella pneumophila), Lyme disease (Borrelia burgdorferi), peptic ulcers (Helicobacter pylori), gonorrhea (Neisseria gonorrhoeae), and Mycobacterium avium in AIDS patients. They are also effective against skin and soft tissue pathogens (staphylococci, Propionibacter acne, and Streptococcus pyogenes). They are all effective orally vs. Gram-positive bacteria and some Gram-negative organisms such as Hemophilus influenzae and Mannheimia spp. All macrolides inhibit protein synthesis.
Macrolides being used for other uses include immunosuppressants such as sirolimus (rapamycin), and tacrolimus (FK506), antiparasitics such as avermectin, and antitumor agents such as the epothilones. The biosynthesis of polyketide macrolides has been subjected to genetic engineering [83, 123], and combinatorial biosynthesis has become very important [119] as it has for cyclic lipopeptide antibiotics.
Peptides are an important part of the antibiotic area [8, 52]. They include vancomycin, teicoplanin, the streptogramins, and the bacteriocins. Streptogramins include pristinamycin, and the virginiamycin M and virginiamycin S pair, which act synergistically and are produced by Streptomyces virginiae. The glycopeptide vancomycin was for years the molecule of choice to treat infections caused by antibiotic-resistant bacteria. However, over the years, resistance to vancomycin developed, especially in the case of infections by VRE. This problem has been combated by the use of the related lipoglycopeptide teicoplanin (Targocid) as well as daptomycin, linezolid, synercid, and televancin. Teicoplanin also has fewer side-effects than vancomycin and a longer half-life in the human body.
Lipophilic analogs of teicoplanin and ristocetin have improved antibiotic activity. One has antiviral activity vs. influenza [89]. Song [103] described five antimicrobials which could replace vancomycin and new carbapenems active against Gram-negative infections. They are the glycopeptides dalbavancin, televancin and oritavancin, the lipopeptide daptomycin, the cephalosporins ceftobiprole and ceftaroline and the diaminopyrimidine iclaprim. Dalbavancin is a semi-synthetic lipoglycopeptide derived from teicoplanin. Televancin is a lipoglycopeptide that inhibits cell wall formation and disrupts membrane barrier function. Ceftobiprole is a cephalosporin that has both Gram-positive and Gram-negative antibacterial activity. Iclaprim is an inhibitor of dihydrofolate reductase that inhibits Gram-positive and Gram-negative bacteria. Gram-negative and Gram-positive activity is also shown by doripenem, a carbapenem approved by FDA in 2007.
More than 1,000 antimicrobial peptides are known. Bacteriocins are ribosomally synthesized antimicrobial peptides and are divided into different groups. One group (the cationic peptide type A1 antibiotics) is made up of the lantibiotics, which contain unusual amino acids, one of which is lanthionine. More than 50 lantibiotics produced by Gram-positive bacteria are known. Type A(1) lantibiotics are potent and broad- spectrum, have low toxicity, and are active in vivo [6]. They include nisins A and Z, subtilin, nukacin ISK-1, several lacticins, mersacidin, actagardine, cinnamycin, SapB, sublancin, gallidermin, plantaricin W, mutacin and epidermin. Some combinations work synergistically. The most well known lantibiotic, nisin, was discovered in the 1920s and has been used as a food preservative for more than 40 years. It has no human toxicity but is broken down in the gastrointestinal tract and has low stability at physiological pH levels. Nisin inhibits bacterial peptidoglycan synthesis and produces membrane pores by reacting with the cell wall precursor lipid II. It is active against bacterial mastitis, oral decay, enterococcal infection, peptic ulcers, and enterocolitis. It is also reported to inhibit experimental vascular graft infection by methicillin-resistant Staphylococcus epidermidis. Mersacidin and actagardine inhibit methicillin-resistant Staphylococcus aureus (MRSA), bacterial mastitis, oral decay and acne. Gallidermin and epidermin are effective against acne, eczema, folliculitis, and impetigo. Lacticin 3147 works against bacterial mastitis, MRSA, enterococci, and acne. Cinnamycin acts against inflammation and viral infection. Despite these important abilities, nisin and other lantibiotics have not been extensively used for therapy [101]. Apparently, their manufacture is both time-consuming and expensive, and the high cost limits their use. Furthermore, since they are administered by injection rather than orally, their production is subject to intensive regulation concerning sterility.
In the search for new antibiotics, many of the new products are made by chemists by modification of natural antibiotics; this process is called “semi-synthesis”. As early as 1974, over 20,000 semi-synthetic penicillins, 4,000 cephalosporins, 2,500 tetracyclines, 1,000 rifamycins, 500 kanamycins, and 500 chloramphenicols had been prepared.
Completely synthetic antimicrobials include the quinolone and fluoroquinolone groups, discovered as inhibitors of DNA gyrase. They are related to the structure of a natural product, the alkaloid quinine [81]. The quinolone era started with the discovery of chloroquine (called Resoquine) for malaria at the Bayer company in the early 1930s. It was followed in 1962 at the Sterling-Winthrop Research Institute in New York by Lesher and coworkers who synthesized nalidixic acid (Negram®), a 4-quinolone with strong Gram-negative antibacterial activity [108]. Additional quinolones were developed in 1968. These included cinoxacin, rosoxacin, pipemidic acid, piromidic acid, and oxolinic acid.
Work on the fluoroquinolones was first done at Riker Laboratories in Minnesota in the mid-1970s. It was discovered that fluorine at C6 markedly improved activity of the quinolones and thus the commercial fluoroquinolones were born. In 1981, the Merck company reported on their 6-fluoroquinolone, norflacin, which was licensed from Kyorin Pharmaceuticals. This was followed by development of ciprofloxacin (Cipro®), ofloxacin, enoxacin, perfloxacin, moxifloxacin and levofloxacin. Ciprofloxacin, developed at Bayer, became very successful due to its high activity. It was approved by FDA as an oral drug in 1987 and as an IV drug in 1991.
Of the 25 top-selling drugs in 1997, 42 % were natural products or derived from natural products [21]; of these, antibiotics contributed 67 % of sales. The worldwide market for antibiotics is $35 billion. If one includes antiviral agents, the figure reached $55 billion in 2000. Antibiotics from species of Streptomyces alone had a market of $25 billion in 2001 [61]. The market for antifungal drugs in 2002 reached $4 billion [32]. At their peaks, individual groups of antibiotics reached impressive sales figures. β-Lactam antibiotics constituted a major part of the market: cephalosporins sold for $11 billion, penicillins for $8 billion, and carbapenems and other β-lactams for $3 billion, making a total of around $22 billion. Sales of macrolides reached $7 billion, mainly involving tylosin, clarithromycin, azithromycin and erythromycin. Aminoglycoside sales reached $1.8 billion and tetracycline sales reached $1.4 billion. Combined sales of the glycopeptides vancomycin and teicoplanin were $1 billion. The market for all quinolones amounted to $6.4 billion with fluoroquinolones accounting for $3.2 billion, dominated by levofloxacin (Levaquin®). Markets for the synthetic azoles reached $2 billion [53]. Global sales of oral antibiotics amounted to $25 billion in 2005 [31]. The antiviral market was $16 billion.
Individual antimicrobials with annual markets over $1 billion dollars include augmentin, a combination of a semi-synthetic penicillin and the β-lactamase inhibitor, clavulanic acid ($2.1 billion), the quinolones ciprofloxacin ($1.8 billion) and levofloxacin/ofloxacin ($1.1 billion), the semi-synthetic macrolides azithromycin (Zithromax®; $2 billion) and clarithromycin (Biaxin®; $1.6 billion) and the semi-synthetic cephalosporin ceftriazone (Rocephin®; $1.1 billion) [107, 119].
Worldwide antibiotic production amounts to about 100,000 tons. Included are 60,000 tons of penicillins, 5,500 tons of tetracyclines, 2,500 tons of cephalosporins. Antibiotics that are natural or derived from natural products include β-lactam antibiotics such as ampicillin (5,000 tons per year), cephalexin (4,000 tons), amoxicillin (16,000 tons), and cefadroxil (1,000 tons). Macrolides at high tonnage include azithromycin (1,500 tons) and clarithromycin (1,500 tons). Glycopeptides such as vancomycin and teicoplanin are produced at a total of 9,000 tons.
Anticancer agents
An extremely important concept for the further development of natural products is that compounds which possess antibiotic activity also possess other activities. Some of these activities had been quietly exploited in the past, and it became clear in the 1980s that such broadening of scope should be expanded. Thus, a broad screening of antibiotically active molecules for antagonistic activity against organisms other than microorganisms, as well as for activities useful for pharmacological or agricultural applications, was pursued in order to yield new and useful lives for “failed antibiotics.” This resulted in the development of a large number of simple in vitro laboratory tests, e.g., enzyme inhibition screens to detect, isolate and purify useful compounds. Fortunately, we entered into a new era in which microbial metabolites were applied to diseases heretofore only treated with synthetic compounds, i.e., diseases not caused by bacteria and fungi and huge successes were achieved. An area that experienced great success was that of antitumor agents. Of the 140 anticancer agents approved since 1940 and available for use, over 60 % can be traced to a natural product. Of the 126 small molecules among them, 67 % are natural in origin [77]. In 2000, 57 % of all drugs in clinical trials for cancer were either natural products or their derivatives [33].
In their review on the use of microbes to prescreen potential antitumor compounds, Newman and Shapiro [78] concluded that microorganisms have played an important role in identifying compounds with therapeutic benefit against cancer. Most of the important compounds used for chemotherapy of tumors are microbially produced antibiotics. Approved antitumor agents from microorganisms include actinomycin D (dactinomycin), anthracyclines, including daunorubicin, doxorubicin (adriamycin), epirubicin, pirarubicin, idarubicin, valrubicin and amrubicin, glycopeptides (bleomycin, phleomycin), the mitosane mitomycin C, and the anthracenones (mithramycin, streptozotocin, pentostatin).
A modified anthracycline, 11-hydroxyaclacinomycin A, was produced by cloning the doxorubicin resistance gene and the aklavinone 11-hydroxylase gene dnrF from the doxorubicin producer, Streptomyces peucetius subsp. caesius, into the aclacinomycin A producer. The hybrid molecule showed greater activity against leukemia and melanoma than aclacinomycin A. Another hybrid molecule produced was 2′-amino-11-hydroxyaclacinomycin Y, which was highly active against tumors. Additional new anthracyclines have been made by introducing DNA from Streptomyces purpurascens into Streptomyces galilaeus, both of which normally produce known anthracyclines.
Novel anthracyclines were produced by cloning DNA from the nogalomycin producer, Streptomyces nogalater, into Streptomyces lividans and into an aclacinomycin-negative mutant of S. galilaeus. Cloning of the actI, actIV, and actVII genes from S. coelicolor into the 2-hydroxyaklavinone producer, S. galilaeus 31,671 yielded novel hybrid metabolites, desoxyerythrolaccin and 1-O-methyl-desoxyerythrolaccin. Similar studies yielded the novel metabolite aloesaponarin II. Epirubicin (4′-epidoxorubicin) is a semi-synthetic anthracycline with less cardiotoxicity than doxorubicin. Genetic engineering of a blocked S. peucetius strain provided a new method to produce it. The gene introduced was avrE of the avermectin-producing S. avermitilis or the eryBIV genes of the erythromycin producer, Saccharopolyspora erythraea. These genes and the blocked gene in the recipient are involved in deoxysugar biosynthesis.
An unusual source of secondary metabolites is the myxobacteria, relatively large Gram-negative rods that move by gliding or creeping. They form fruiting bodies and have a very diverse morphology. Over 400 compounds had been isolated from these organisms by 2005 but the first in clinical trials were the epothilones, potential antitumor agents that act like taxol but are active vs. taxol-resistant tumors. They are 16-member ring polyketide macrolide lactones produced by the myxobacterium Sorangium cellulosum, which were originally developed as antifungal agents against rust fungi [54], but have found their use as antitumor compounds [56]. They contain a methylthiazole group attached by an olefinic bond. They are active against breast cancers, including those that are resistant to taxol and other forms of chemotherapy. They bind to and stabilize microtubules essential for DNA replication and cell division, even more so than taxol. One epothilone, ixabepilone, produced chemically at Bristol Myers-Squibb from epothilone B, was approved by FDA. By preventing the disassembly of microtubules, epothilones cause arrest of the tumor cell cycle at the GM2/M phase and induce apoptosis (programmed cell death). The mechanism is similar to that of taxol but epothilones bind to tubulin at different binding sites and induce microtubule polymerization. Production of epothilone B by Sorangium cellulosum is accompanied by the undesirable epothilone A. Production of B over A was favored by adding sodium propionate to the medium. Epothilone polyketides are more water-soluble than taxol. The producing microbe is a very slow grower (16-h doubling time) and low producer (20 μg/ml).
Plants have been a useful source of anticancer agents. Etoposide and teniposide were derived as semi-synthetic derivatives of podophyllotoxin, an antimitotic metabolite of mayapple roots [41]. The mayapple plant is an old herbal remedy. Etoposide is a topoisomerase II inhibitor. This essential enzyme is involved in eukaryotic cell growth by regulating levels of DNA supercoiling [9]. Vinca alkaloids, such as vinblastine and vincristine, originate from the Madagascar periwinkle plant. The naphthoquinone pigment shikonin is produced by cell culture of the plant Lithospermum erythrorhizon, a herbal medicine remedy. Shikonin and two derivatives inhibit tumor growth in mice bearing Lewis lung carcinoma [68]. Other promising plant products include curcumin, resveratrol, gingrerole, capsaicin, epogallocatechin gallate, genistein, flapopiridol, and silymarin.
Taxol (paclitaxel) has been a very successful antitumor molecule. It was originally discovered in plants but has also been found to be a fungal metabolite [104]. This diterpene alkaloid was approved for breast and ovarian cancer and acts by blocking depolymerization of microtubules. In addition, taxol promotes tubulin polymerization and inhibits rapidly dividing mammalian cancer cells. Taxol was originally isolated from the bark of the Pacific yew tree (Taxus brevifolia) but it took six trees of 100 years of age to treat one cancer patient. It is now produced by plant cell culture or by semi-synthesis from taxoids made by Taxus species. These species make more than 350 known taxoid compounds. Early genetic engineering of S. cerevisiae yielded no taxadiene (the taxol precursor) because too little of the intermediate, geranylgeranyl diphosphate, was formed. When the Taxus canadensis geranylgeranyl diphosphate synthase gene was introduced, 1 mg/l of taxadiene was obtained [36]. More recent metabolic engineering studies [50] yielded a Saccharomyces cerevisiae strain producing over 8 mg/l taxadiene and 33 mg/l geranyl geraniol. The use of cells of the plant Taxus chinensis to produce taxol became the industrial means to make the compound. The addition of methyl jasmonate, a plant signal transducer, increased production from 28 to 110 mg/l. The optimum temperature for growth of T. chinensis is 24 °C and that for taxol synthesis is 29 °C. Shifting from 24 to 29 °C at 14 days gave 137 mg/l at 21 days [30]. There is a 6 week process yielding 153 mg/l with Taxus sp [18]. Taxol has sales of $1.6 billion per year. Fungi such as Taxomyces adreanae, Pestalotiopsis microspora, Tubercularia sp., and Phyllosticta citricarpa produce taxol [104, 106, 116] but the production level is low, e.g., 265 μg/l produced by P. citricarpa [66]. Taxol has antifungal activity by the same microtubule mechanism, especially against oomycetes [105]. Oomycetes are water molds exemplified by plant pathogens such as Phytophthora, Pythium, and Aphanomyces.
Camptothecin is a modified monoterpene indole alkaloid produced by certain plants (angiosperms) [115]. It also is produced by an endophytic fungus (Entrophospora infrequens) from the plant Nathapodytes foetida. It is used for recurrent colon cancer and has unusual activity against lung, ovarian, and uterine cancers [5]. Colon cancer is the second-leading cause of cancer fatalities in the USA and the third most common cancer among US citizens. Camptothecin is known commercially as Camptosar (Pharmacia) and Campto (Aventis and Yakult) and achieved sales of $1 billion in 2003 [70]. Its water-soluble derivatives irinotecan and topotecan are used clinically. In view of the low concentration of camptothecin in tree roots and poor yield from chemical synthesis, the fungal fermentation is very promising for industrial production of camptothecin. Its cellular target is type I DNA topoisomerase. When patients become resistant to irenotecan, its use can be prolonged by combining it with the monoclonal antibody Erbitux (Cetuximab®) from ImClone/BMS. Erbitux blocks a protein that stimulates tumor growth and the combination helps metastatic colorectal cancer patients expressing epidermal growth factor receptor (EGFR). This protein is expressed in 80 % of advanced metastatic colorectal cancers. The drug combination reduces invasion of normal tissues by tumor cells and the spread of tumors to new areas.
Metastatic testicular cancer, although rather uncommon (1 % of male malignancies in the USA; 80,000 in the year 2000 as compared to 190,000 cases of prostate cancer), is the most common carcinoma in men aged 15–35. It is quite interesting that the cure rate for disseminated testicular cancer was 5 % in 1974; later, it rose to 90 % mainly due to combination chemotherapy with the natural products bleomycin and etoposide and the synthetic cisplatin [46].
Angiogenesis (recruitment of new blood vessels) is necessary for tumors to obtain oxygen and nutrients. Tumors actively secrete growth factors that trigger angiogenesis. The concept of angiogenesis was established by Prof. Judah Folkman [25]. He proposed that tumor growth depends on angiogenesis and proposed the use of angiogenesis inhibitors as antitumor agents, i.e., to target activated endothelial cells. He further proposed that the vascular endothelial growth factor (VEGF) is involved in angiogenesis and that it could be a target for anti-angiogenic drugs. Fumagillin, produced by Aspergillus fumigatus, was one of the first agents found to act as an anti-angiogenesis compound. Next to come along for angiogenesis inhibition were its oxidation product ovalacin and the fumagillin analogue TNP470 (=AGM-1470). TNP470 binds to and inhibits type 2 methionine aminopeptidase (MetAP2). This interferes with amino-terminal processing of methionine, which may lead to inactivation of enzymes essential for proliferation and in vitro migration of endothelial cells. In animal models, TNP470 effectively treated many types of tumor and metastases.
Fumagillin also has immunosuppressive activity that is correlated with its anti-angiogenic activity. MetAP2 is also the fumagillin target in yeast, which makes MetAP2 and another similar enzyme MetAP1. Wild-type yeast expressing both map1 and map2 genes are insensitive whereas map1 mutants are sensitive to fumagillin and ovalicin; map2 mutants are insensitive. The primary function of these two enzymes is to remove the initiator methionine at the amino terminus of proteins. This posttranslational processing step is necessary for the myristoylation, which is required for protein targeting and stability of certain proteins. N-myristoylation is essential for viability of S. cerevisiae and Candida neoformans. Differences between substrate specificity of the C. neoformans enzyme and the mammalian enzyme exists which suggests the possibility of developing novel antifungal agents [27]. Inhibition of MetAP2 might be expected to make short-lived enzymes more stable to desirable degradation and this might inhibit angiogenesis. Fumagillin is active against Nosia sp, a fungus causing disease of honeybees. Also, TNP470 is potent against microsporidia, obligate intracellular parasites causing diarrhea and wasting syndrome in immunocompromised patients including those with AIDS.
The first anti-angiogenesis drug on the market for cancer was Avastin (bevacizumab), a monoclonal antibody from Genentech/Roche. It acts against the vascular endothelial growth factor (VEGF), an angiogenic factor, and is used for metastatic colorectal cancer. Additional FDA-approved angiogenesis inhibitors are pegaptanib (Macugen®) and ranibizumab (Lucentis®). Macugen is an aptamer of VEGF and Lucentis is an anti-VEGF antibody. By 2008, ten anti-angiogenesis drugs had been approved. Eight are used against cancer and two are employed for treatment of age-related macular degeneration. Anti-angiogenesis therapy is now known as one of four cancer treatments. The other three are surgery, radiotherapy, and chemotherapy. By the end of 2007, 23 anti-angiogenic drugs were in Phase III clinical trials and more than 30 were in Phase II.
Genome mining is useful for identifying genetic units with potential for synthesizing new drugs [97, 118]. As a result of such an effort with Micromonospora sp., a new antitumor drug was discovered [127]. The compound, ECO-04601, is a farnesylated dibenzodiazapene that induces apoptosis.
An analog of the immunosuppressive agent rapamycin with antitumor activity is temsirolimus. It inhibits mTOR, which is a kinase. The drug, produced by Wyeth, was approved for renal cell carcinoma [93].
The marine environment offers new opportunities for the discovery of antitumor agents [63]. The genus Salinospora and its 2 species, S. tropica and S. areniola, have been isolated around the world, S. tropica makes a bicyclic beta-lactone gamma-lactam called salinosporamide A which is a proteasome inhibitor and has antitumor activity. Also the genus Marinophilus contains species that produce novel polyenes with potent antitumor activity. The symbionts of marine invertebrate animals continue to reveal interesting natural products [43]. Variants of the toxic dolastin from the sea hare Dolabella auricalaria seem promising against cancer. These include soblidotin (T2F 1027) which completed Phase II against soft tissue sarcoma, and synthadotin (=tasidotin = 1LX 651), which is at the same clinical stage against melanoma, prostate and non-small cell lung cancers. These are thought to be produced by cyanobacteria sequestered by the marine invertebrates in their diet.
A promising drug for clinically reversing multidrug resistance in tumor cells is the non-immunosuppressive cyclosporin A derivative valspodar (PSC-833) of Novartis [94].
Immunosuppressants
Cyclosporin A was originally discovered as a narrow spectrum antifungal peptide produced by the mold, Tolypocladium nivenum (previously Tolypocladium inflatum). Discovery of its immunosuppressive activity led to its use in heart, liver, and kidney transplants and to the overwhelming success of the organ transplant field. Sales of cyclosporin A reached $1 billion in 1994. Although cyclosporin A had been the only product on the market for many years, two other products, produced by actinomycetes, provided new opportunities. These are rapamycin (=sirolimus) [113] and the independently discovered tacrolimus (=FK506, Fujimycin). They are both narrow spectrum polyketide antifungal agents, which are 100-fold more potent that cyclosporin as immunosuppressants and less toxic. All three immunosuppressants were originally described as antifungal agents. Non-immunosuppressive derivatives have been developed with good antifungal activity [34]. Tacrolimus and rapamycin have both been used clinically for many years. Tacrolimus was almost abandoned by the Fujisawa Pharmaceutical Co. after initial animal studies showed dose-associated toxicity. However, Dr. Thomas Starzl of the University of Pittsburgh, realizing that the immunosuppressant was 30 to 100-fold more active than cyclosporin tried lower doses which were very effective and non-toxic, thus saving the drug and many patients after that, especially those that were not responding to cyclosporin [4]. Since its introduction (1993 in Japan; 1994 in USA), tacrolimus has been used for transplants of liver, kidney, heart, pancreas, lung, intestines and for prevention of graft-versus-host disease. Recently, a topical preparation has been shown to be very active against atopic dermatitis, a widespread skin disease. Tacrolimus had a market of $2 billion in 2007.
Rapamycin does not exhibit the nephrotoxicity of cyclosporin A and tacrolimus and is synergistic with both compounds in immunosuppressive action [95]. By combining it with either, kidney toxicity is markedly reduced. Rapamycin has been the basis of chemical modification to yield important products such as everolimus, temsirolimus (CCI-779) and deforolimus (A23573). Rapamycin is not only an immunosuppressant but also has antifungal, antitumor, neuroprotective, autoimmune and anti-aging properties. Its biosynthesis, regulation and mutagenic improvement of production have been reviewed by Park et al. [84].
Studies on the mode of action of these immunosuppressive agents have markedly expanded current knowledge of T cell activation and proliferation. Rapamycin, tacrolimus and cyclosporin A all act by interacting with an intracellular protein (an immunophilin) thus forming a novel complex which selectively disrupts signal transduction events of lymphocyte activation. By binding to its immunophilin (FKBP), rapamycin inhibits a unique growth regulation path utilized by lymphocytes in responding to several cytokines. The targets of cyclosporin A, tacrolimus and sirolimus are inhibitors of signal transduction cascades in microorganisms and humans. In humans, the signal transduction pathway is required for activation of T cells. The targets are highly conserved from microbial eukaryotes to humans. When these compounds enter cells, they form complexes with their immunophilins and inhibit the latter’s prolyl isomerase activity, usually involved in protein folding, but this is not the crucial step. The combination of cyclosporin or tacrolimus and their specific immunophilin inhibits calcineurin, a serine-threonine-specific protein phosphatase that is normally activated by calmodulin in response to increases in intracellular Ca2+. A previously unknown protein called mTOR (a member of the family of lipid/protein kinases) is part of the sirolimus-sensitive signal transduction pathway. Rapamycin combined with its immunophilin inhibits TOR kinase which normally transduces growth promoting signals that are sent in response to nutrients (e.g., amino acids) and growth factors.
TOR has phosphatidylinositol lipid kinase activity, which is involved in cell cycle regulation. TOR proteins of yeast and mammals share sequence similarity to protein and lipid kinases although their predominant activity is thought to be that of phosphatidylinositol lipid kinase. Many homologues exist in yeast and mammalian cells (called RAFT, FRAP, mTOR, SEP, RAPT1). TORs respond to nitrogen sources and other growth factors to regulate translation, transcription and cell cycle progression. The sensitivity of yeast to cyclosporin A and tacrolimus is due to the need for calcineurin to promote yeast survival during cation stress. In Cryptococcus neoformans, calcineurin is needed for virulence. Other fungi inhibitable by cyclosporin and tacrolimus are Coccidiodes immitis, Aspergillus niger, A. fumigatus and Neurospora crassa, suggesting that calcineurin may be necessary for viability in these species. Two non-immunosuppressive analogs of cyclosporin are active against Cryptococcus neoformans [34]. Like the immunosuppressive cyclosporin, they act by binding to cyclophilin A and inhibiting the action of the fungal calcineurin. A non-immunosuppressive tacrolimus derivative, a C18 hydroxy C21 ethyl analogue called L-685,818, inhibits C. neoformans by inhibition of calcineurin. It is non-immunosuppressive because its combination with human immunophilin (human FKBP12) does not inhibit vertebrate calcineurin but when in combination with fungal FKBP12, it does. Thus, it is possible to exploit subtle differences in the structures of human and fungal FKBP12. Since TOR proteins are involved negatively in nutrient repression, addition of rapamycin induces certain yeast genes whose transcription is repressed by nitrogen abundance, glucose abundance and also genes involved in the diauxic shift [27]. Rapamycin and its less immunosuppressive analogs are effective against Candida albicans and C. neoformans via FKBP12-dependent inhibition of TOR kinases [35]. These enzymes are necessary for stationary phase entry, expression of ribosomal protein genes, nitrogen catabolite repression and translation.
Rapamycin also has antitumor activity. Whereas cyclosporin A promotes tumor growth and many transplant patients are killed by tumors, rapamycin inhibits tumor growth by interfering with angiogenesis and also is an inducer of apoptosis [57, 90]. Rapamycin is also able to reverse multidrug resistance to antitumor agents in mammalian cells. Cyclosporin and tacrolimus also have this ability. In addition to their actions as immunosuppressants, antitumor agents and antifungal agents, the ascomycins, structurally related to rapamycin, have anti-inflammatory action and are being used for topical treatment of skin diseases such as atopic dermatitis, allergic contact dermatitis and psoriasis [28]. Tacrolimus is also being studied for skin diseases.
Cyclosporin A has activity against the malaria parasite Plasmodium falciparum in agreement with its genome containing sequences encoding cyclophilin and calcineurin [42]. Another activity of tacrolimus and rapamycin is stimulation of nerve cells [55]. They thus might find use for combating neurological disorders. Certain ascomycin derivatives made by combinatorial biosynthesis are being studied for nerve regeneration [92]. Cyclosporin A analogs are being clinically tested against the inflammatory disease asthma and have shown promising results [45]. They exhibit decreased nephrotoxicity and have different pharmacology and metabolism.
A very old broad-spectrum antibiotic compound, mycophenolic acid, has an amazing history. Bartolomeo Gosio (1863–1944), was an Italian physician who discovered the compound in 1893 [10]. Gosio isolated a fungus from spoiled corn which he named Penicillium glaucum, which was later reclassified as Penicillium brevicompactum. He isolated crystals of the compound from culture filtrates in 1886 and found them to inhibit growth of Bacillus anthracis. This was the first time an antibiotic had been crystallized and the first time that a pure compound had ever been shown to have antibiotic activity. The work was forgotten but fortunately the compound was rediscovered by Alsberg and Black [3] and given the name mycophenolic acid. They used a strain originally isolated from spoiled corn in Italy called Penicillium stoloniferum, a synonym of P. brevicompactum. The chemical structure was elucidated many years later by workers in England [12]. Mycophenolic acid has antibacterial, antifungal, antiviral, antitumor, antipsoriasis and immunosuppressive activities. It was never commercialized as an antibiotic because of its toxicity, but its 2-morpholinoethylester was approved as a new immunosuppressant for kidney transplantation in 1995 and for heart transplants in 1998. The ester is called mycophenolate mofetil (CellCept®) and is a prodrug that is hydrolyzed to mycophenolic acid in the body.
Immunosuppressants, such as cyclosporin A and tacrolimus, convert the normally fungistatic activity of azoles (i.e., fluconazole) against C. albicans, Candida glabrata and Candida krusei into fungicidal activity [80]. They do this by inhibiting the protein phosphatase calcineurin. Even non-immunosuppressive analogs of tacrolimus have this ability. Non-azole drugs that inhibit other steps of ergosterol biosynthesis (terbinafine, fenpropimorph) are also improved in activity by immunosuppressants and their non-immunosuppressive analogs.
Rapamycin extends life span in mice and is being considered for possible use against progeria (Hutchinson–Gilford progeria syndrome) in children. This is a rare disease that resembles accelerated aging and kills children in their teens. Rapamycin, when tested on cells from children suffering with progeria, promoted cleavage of progerin, the mutant protein that accumulates in cells of affected children, and it extended survival of the cells [26].
Prodigiosins, red microbial pigments produced by Serratia marcescens, have immunosuppressive and anticancer activities [82]. Pigments produced by Monascus have antibiotic, immunosuppressive, and hypotensive activities.
Hypocholesterolemic agents
Only 30 % of the cholesterol in the human body comes from the diet. The remaining 70 % is synthesized by the body, mainly in the liver. Many people cannot control their cholesterol at a healthy level by diet alone but must depend on hypocholesterolemic drugs. The statins inhibit de novo production of cholesterol in the liver, the major source of blood cholesterol. High blood cholesterol leads to atherosclerosis, which is a causal factor in many types of coronary heart disease, a leading cause of human death. Statins were a success because they reduced total plasma cholesterol by 20–40 % whereas the previously used fibrates only reduced it by 10–15 % [65]. The statins are microbially produced enzyme inhibitors, inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A reductase, the regulatory and rate-limiting enzyme of cholesterol biosynthesis in liver.
The discovery and developments of the statins present a fascinating story. Their history has been described by Akira Endo, the discoverer of the first statin, compactin [48]. They were first discovered in fungi in England and Japan in 1975–6. Compactin had been reported as an antifungal agent by Brown et al. [20] of Glaxo from P. brevicompactum. Earlier, in Japan, the Sankyo pharmaceutical company had supported Endo for a sabbatical leave at Albert Einstein Medical College (NYC) to study phospholipid metabolism under Bernard Horecker. Upon returning in 1968 to Sankyo, Endo screened 6,000 fungal extracts for inhibition of cholesterol biosynthesis by rat liver membranes and found two actives, i.e., ML-236A and B, produced by Penicillium citrinum. ML-236B was compactin (=mevastatin). It was found to inhibit 3-hydroxy-3-methylglutaryl coenzyme A reductase [49]. In 1976, Sankyo prepared a patent application on compactin. However, Sankyo did not commercialize compactin and it never became a commercial drug. In 1976, H. Boyd Woodruff, who was Merck’s representative in Japan, heard of the work of Endo, and requested a sample of compactin. The two companies signed a confidentiality agreement and the compactin sample was given to Merck. Merck did tests in cultured mammalian cells, rats and dogs, and, by 1978, had obtained promising results. At that time, the group of Alberts at Merck started to screen for new inhibitors and discovered lovastatin produced by Aspergillus terreus [2], which they called Mevacor®. It had a structure similar to compactin but contained a methyl group. At about the same time, Endo [47] reported the discovery of lovastatin from Monascus ruber and named it monocolin K (=mevinolin). It was patented in Japan but without structural elucidation. Merck filed for a patent containing their findings and the structure of lovastatin. The company received a US patent in 1980 and lovastatin became the first statin on the market. Sankyo, in their tests with compactin on dogs, apparently noted intestinal tumors and in 1980, stopped further testing. Merck also stopped testing at that time and this inactivity existed for 2–3 years. However, since Merck had seen no such tumors with lovastatin, they decided to resume their efforts. Further clinical tests on lovastatin went into full speed in 1982, and the drug was finally approved by FDA in 1987, after clinical tests in humans had shown a lowering of total blood cholesterol of 18–34 %, a 19–39 % decrease in low-density lipoprotein cholesterol (“bad cholesterol”) and a slight increase in high-density lipoprotein cholesterol (“good cholesterol”).
Merck later produced simvastatin (Zocor®) in which the 2-methylbutanoate side-chain of lovastatin was chemically modified to 2,2-dimethylbutanoate; it was launched in 1988. Although compactin was never used medically, Sankyo devised a bioconversion of it by hydroxylation yielding pravastatin which became commercial in 1989 and was then licensed to Bristol-Myers Squibb. The bioconversion was carried out using actinomycetes [87, 100].
In 1985, a 28-year-old postdoctoral associate in the University of Rochester’s Chemistry Department, Bruce D. Roth, was able to chemically synthesize one of the statins that Endo had isolated from his fungus in the 1970s. Two years later, he headed an 18-person group at the Parke-Davis company (now Pfizer) working on synthesis of a synthetic statin called atorvastatin (Lipitor®). They compared atorvastatin in a further clinical trial vs. fluvastatin, lovastatin, pravastatin, and simvastatin; atorvastatin showed the best results. The FDA approved it in January of 1997. Parke-Davis decided to co-market it with Pfizer in 1996. By mid-1998, atorvastatin had 18 % of the statin market as compared to simvastatin’s 37 %. Pfizer, in 2000, purchased Warner-Lambert, the parent of Parke-Davis and became the sole owner of atorvastatin, which became the leading drug in the world.
The largest segment of the pharmaceutical business is for cholesterol-lowering drugs, amounting to about 30 % of the market. Simvastatin reached a market of over $7 billion. Pravastatin attained sales of $5 billion. Atorvastatin became the leading drug in the world at $12 billion per year. A useful review on the statins was published by Manzoni and Rollini [71].
Natural statins are produced by many fungi: A. terreus and species of Monascus, Penicillium, Doratomyces, Eupenicillium, Gymnoascus, Hypomyces, Paecilomyces, Phoma, Trichoderma, and Pleurotis. Although pravastatin is commercially made by bioconversion of compactin, certain strains of Aspergillus and Monascus can produce pravastatin directly [72]. Basidiomycetes, such as Pleurotus ostreatus, also produce lovastatin but at low levels [1].
Simvastatin has traditionally been made by synthetic multistep processes starting with lovastatin. This can now be avoided by use of an Escherichia coli strain over-expressing lovD in the presence of a cell-membrane permeable thioester, i.e., dimethylbutyryl-5-methylmercaptopropionate [121, 122]. The whole-cell procedure converts monocolin J acid to simvastatin acid in high yields, i.e., over 99 %.
Statins reduce cardiovascular events including myocardial infarction, stroke, and death [112]. Not only are they active against arthrosclerosis, the most common cause of death in Western countries, but also improve endothelial function, and have anti-inflammatory, anti-atherothrombosis, immunomodulation, and anti-migration activities. The inflammatory effect is over and above their action in lowering cholesterol [126]. Statins reduce total and LDL-cholesterol and increase HDL-cholesterol. They also reduce the occurrence of Alzheimer's disease. Statins also lower elevated C-reactive protein (CRP) levels independent of their effect on cholesterol [29]. This is important since half of all myocardial infarctions occur in patients with normal LDL levels. High CRP is associated with inflammatory response in atherosclerosis and is a predictor of future cardiovascular mortality. Statins can also prevent stroke, reduce development of peripheral vascular disease. They are also showing beneficial effects for multiple sclerosis and cancer. Experiments with oral statins showed efficacy in a mouse model of multiple sclerosis. The effect appears to be independent of cholesterol lowering. Other activities being studied are stimulation of bone formation and antioxidation [120].
Red yeast has been used as a traditional Chinese food and medicine since 800 a.d. It is also known as red koji or Hongqu. The fermentation organism is Monascus purpureus whose pigments give the food its characteristic color. The organism is used for rice, red wine, red soy bean cheese, meat, and fish, and is authorized for food use in China and Japan. The orange pigments, monascorubin and rubropunctatin, have both antibacterial and antifungal activities [73]. Li Shizhen, the noted pharmacologist of the Ming Dynasty (1368–1644), reported the favorable effects of red rice on blood circulation. More recent work has shown that red rice lowers blood-lipid levels due to its content of statins and clinical trials demonstrated the lowering of cholesterol in humans.
Additional applications of natural products
Inhibitors of angiotensin-converting enzyme (ACE), which are widely used for hypertension and congestive heart failure, are chemicals based on peptides isolated from snake venom [85].
The predecessor of aspirin has been known as far back as the fifth century b.c., at which time it was extracted from willow tree bark by Hippocrates. It probably was used even earlier in Egypt and Babylonia for fever, pain, and childbirth. Such salicylic acid derivatives have been found in plants such as white willow, wintergreen and meadowsweet. Synthetic salicylates were produced on a large scale in 1874 by the Bayer company in Germany.
Natural products have been useful in our battles against viruses. d-nucleoside analogs, usually D-ribose derivatives, such as d-ribavirin (Virazole®), are used as enzyme inhibitors against hepatitis C virus [111]. They also include vidarabin (Ara-A), idoxuridine and acyclovir (Zovirax®), acting against DNA synthesis by herpes simplex virus. Acyclovir was isolated from a sponge. l-nucleoside analogs, usually less toxic than the d-forms, include levorine (l-ribavirin), lamivudine (l-b-1,3-oxathiolanylcytosine) and l-2′,3′-dideoxycytidine (l-ddc) against HIV and hepatitis C. Also important are the HIV inhibitors such as azidothymidine (AZT) against reverse transcriptase, and inhibitors of aspartyl protease, HIV entry, cell fusion and integrase. The reverse transcriptase and protease inhibitors that made it to the market were derived from natural product leads screened at the National Cancer Institute [124]. Then, natural product inhibitors of influenza virus neuraminidase (sialidase) came upon the scene. Combinations of the above small molecules are commonly used for antiviral therapy. Recently, larger molecules such as interferon alpha, monoclonal antibodies, polyclonal antibodies, and nucleic acids are becoming important, all of which are related to natural products.
Microbial toxins have also found use. The toxin from Clostridium botulinum, botulinum A (Oculinum®), is used for neck, eye and mouth spasms and as Botox® for reduction of wrinkles.
Natural products are not only of interest as pharmaceuticals, but also include compounds of use in nutrition and as polymers. The annual tonnage of amino acids includes 2.2 million for l-glutamic acid, 1.5 million for l-lysine, 77,000 for l-threonine, and 16,000 for l-phenylalanine. Vitamin C is made at 100,000 tons and vitamin B12 at 12 tons per year. Industrial biotechnology has penetrated the chemical industry in the areas of fine and bulk chemicals, polymers and energy. In certain ways, industrial biotechnology often outperforms conventional chemical technology, e.g., higher reaction rates, increased conversion efficiency, improved product purity, lower energy consumption, and decreased generation of chemical waste [102]. The natural polymer polylactic acid, which is made at 140,000 tons per year, has properties similar to synthetic polyethylene and polypropylene but is much more biodegradable [11]. Microbes are the workhorses of industrial biotechnology because of high synthetic versatility, ease of using renewable raw materials, high reaction rates, rapid growth, and easy modification by genetic means.
Natural products have solved many other problems of our world because of their unique activities. They include anthelmintic agents such as avermectins [24, 79], bioinsecticides (BT toxin, spinosyns, tetranactin, nikkomycins), enzyme inhibitors (lipstain, desferal, acarbose, validamycins, ancovenine, fibrostatin B, phthoxazoline, bialophos, nikkomycin), terpenoids (sterols, gibberellins, carotenoids, lycopene, drimane sesquiterpenes, β-carotene, ubiquinone, artemisinin, amorphadiene, astaxanthin, farnesine, farnesol, isoprene), nutraceuticals (polyunsaturated fatty acids such as docosahexaenoic acid [DHA] and arachidonic acid, prebiotics, coenzyme Q10, omega-3-fatty acids), surfactants (sophorolipids), polysaccharides (pullulan, xanthan, succinoglycan, alginates), bioherbicides, 1,3-propanediol, 1,2-propanediol (propylene glycol), polymeric plastics (poly[3-hydroxybutyrate], polyhydroxyalkanoates, polyacrylamide, polylactic acid), and the very important area of vaccines.
Decrease of new drug discovery by the pharmaceutical industry
The golden age of antibiotic discovery was from 1940 to 1980. New bioactive products from microbes were discovered at an amazing pace, i.e., 200–300 per year in the late 1970s increasing to 500 per year later. Genetics played a great part in the development of useful industrial products from microbes [110]. Production of antibiotics began with penicillin in the late 1940s and proceeded with great success until the 1980s when it became harder and harder to discover new and useful products. The rate of discovery dropped then and slowed down drastically in the 21st century. The major problem has been the loss of interest by the major pharmaceutical companies in these compounds due to the years of preclinical and clinical development required and the short period given to these organizations by governments for sales before patent expiration. Also, the short period of a patient’s use of such products limits their sales as compared to drugs that are taken by patients daily for many years.
The departure of much of the pharmaceutical industry from the drug discovery effort has had its greatest negative effect in the area of antibiotics. New antibiotics are sorely needed because of (a) the development of antibiotic-resistant pathogens, (b) the emergence of over 30 new diseases since 1980 such as AIDS, Ebola virus, Hanta virus, Cryptosporidium, Legionnaires’ disease, Lyme disease, E. coli 0157:H7; (c) the existence of naturally resistant bacteria such as P. aeruginosa causing fatal wound infections, burn infections, and chronic and fatal infections in lungs of cystic fibrosis patients, Stenotrophomonas maltophilia, Enterococcus faecium, Burkholderia cepacia, and Acinetobacter baumanni and (d) the toxicity of some of the current compounds.
In the 1990–2005 period, a number of large pharmaceutical companies emphasized combinatorial chemistry, and departed from the natural product area. Unfortunately, this attempt to replace natural products with synthetic molecules failed because the chemistry did not create sufficiently diverse or pharmacologically active molecules. As a result, the numbers of (a) FDA drug applications, (b) new drug approvals, (c) new and approved active substances, (d) orphan drug applications, and (e) new chemical entities of the pharmaceutical industry markedly decreased since the late 1990s. The exit of many of the major pharmaceutical companies has left much of the discovery efforts to small companies, and the biotechnology industry.
Despite the above problems, development of new antibiotics has continued, albeit at a much slower pace than in the last century. We have seen the (1) appearance of newly discovered antibiotics (e.g., candins), (2) development of old but unutilized antibiotics (e.g., daptomycin, lipiarmycin), (3) production of new semisynthetic versions of old antibiotics (e.g., glycylcyclines, streptogramins), as well as the (4) very useful application of old but underutilized antibiotics (e.g., teicoplanin).
Possible solutions
Research on natural products must continue due to unmet needs. It is obvious that we cannot ignore the opportunities presented to us by natural products. They include their novelty, complexity, remarkable diversity of structures and activities, utility as biochemical probes, existence of novel and sensitive assay methods, improvements in isolation, purification and characterization, and new production methods. The number of chirality centers, bridges, rings, and functional groups in natural products make them much more complex than synthetic compounds. Although combinatorial chemistry has not been of significant use, combinatorial biosynthesis has been helpful in the hunt for new products. This has involved the genetic modification of a producing organism, and/or exchange of genes between organisms to create useful hybrid molecules [8, 60, 74, 119] including new erythromycins, spiramycins, tetracenomycins, anthracyclines and nonribosomal peptides. However, there still is an urgent need for more secondary metabolites to be discovered for use in medicine.
What is needed are the further applications of (1) high-throughput screening to natural product libraries and (2) combinatorial chemistry using natural product scaffolds [37, 64, 114]. There is a higher hit rate in high-throughput screening of natural product collections than of combinatorial libraries of synthetic compounds [16, 17]. Furthermore, the number of compounds in a chemical library is not as important as the biological relevance, design and diversity of the library, and a scaffold from nature provides viable, biologically validated starting points for the design of chemical libraries. It is clear that the role of combinatorial chemistry, like those of structure–function drug design and recombinant DNA technology decades ago, is that of complementing and assisting natural product discovery and development, not replacing them [86]. The combining of natural product screening with high-throughput screening, combinatorial chemistry, genomics, proteomics, metabolomics, and discoveries in biodiversity provide real opportunities for the future [40]. Application of systems biology [96] to antibiotic production is another potential solution.
Opportunity also exists in the deeper examination of the microbial world. Bacteria have existed on earth for over 3 billion years, and eukaryotes have been around for 1 billion years. Since 95–99.9 % of organisms existing in nature have not yet been cultured in the laboratory, only a minor proportion of bacteria and fungi have thus far been examined for secondary metabolite production. It has been estimated that 30 g of soil contains 20,000 common bacterial species and perhaps 500,000 rare species. Another estimate is that that 1 g of soil contains 1,000–10,000 species of undiscovered prokaryotes. About 65,000–70,000 fungal species have been recognized but it has been estimated that from 0.25 to 9.9 million exist [99]. Their total weight is thought to be higher than that of humans. Of the fungal species that have been described, only about 16 % have been cultured. The use of fungal ecology in the search for new drugs is extremely important. The estimated number of fungal species is more than five times the predicted number of plant species and 50 times the estimated number of bacterial species. The well-known concept involving the need for isolation of microbial strains from different geographical and climatic locations around the world, in order to insure microbial diversity in collections, still gathers support. It is obvious that we need to improve our methodology of finding new natural products.
What the government could do
The opportunities described above of combining screening with modern techniques should be seriously considered by the pharmaceutical industry. However, if we ever are to solve the problem of fewer and fewer drugs being commercialized, the government must also contribute. Consider what the pharmaceutical industry has faced in the 21st century [22, 51, 117]: 2–10 years for discovery, 4 years for pre-clinical development, 1 year for clinical development in Phase I, 1.5 years for Phase II, 3.5 years for Phase III, 1 year of FDA review and approval, and 1 year of post-marketing testing. Thus, the total time is 14 years but in certain cases, it has reached 22 years. From 1999 to 2003, the total cost rose from $500–600 million to $900 million. The US government has made an important step in the right direction, i.e., the establishment of its translational center for new drug discovery. However, what is clearly needed is the granting of more time for the drug industry to market their discovered products before they become generic drugs. The time now is 22 years from the time of patent approval and this is clearly too short when one considers the length of time and the funds needed for discovery, pre-clinical and clinical development, etc.
References
Alarcon J, Aguila S, Arancibia-Avila P, Fuentes O, Zamorano-Ponce E, Hernandez M (2003) Production and purification of statins from Pleurotus ostreatus (Basidiomycetes) strains. Z Naturforsch 58:62–64
Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, Rothrock J, Lopez M, Joshua H, Harris E, Patchett A, Monaghan R, Currie S, Stapley E, Albers-Schonberg G, Hensens O, Hirshfield J, Hoogsteen K, Liesch J, Springer J (1980) Mevinolin. A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci USA 77:3957–3961
Alsberg CL, Black OF (1913) Contributions to the study of maize deterioration. Biochemical and toxological investigations of Penicillium puberulum and Penicillium stoloniferum. USDA Bur. Plant Ind., Bull. No. 270, Govt. Printing Office, Washington, DC
Amaya T, Hiroi J, Lawrence ID (2002) Tacrolimus and other immunosuppressive macrolides in clinical practice. In: Omura S (ed) Macrolide antibiotics: chemistry, biology and practice, 2nd edn. Academic/Elsevier, San Diego, pp 421–452
Amna T, Puri SC, Verma V, Sharma JP, Khajuria RK, Spiteller M, Qasi GN (2006) Bioreactor studies on the endophytic fungus Entrophospora for the production of an anticancer alkaloid camptothecin. Can J Microbiol 52:189–196
Asaduzzaman SM, Sonomoto K (2009) Lantibiotics: diverse activities and unique modes of action. J Biosci Bioeng 107:475–487
Baltz RH (2007) Antimicrobials from actinomycetes: back to the future. Microbe 2:125–131
Baltz RH (2012) Combinatorial biosynthesis of cyclic lipopeptide antibiotics: a model for synthetic biology to accelerate the evolution of secondary metabolic biosynthetic pathways for nonribosomal peptides. ACS Synth Biol (dx.doi.org/10.1021/sb3000673)
Bender RP, Jablonksy MJ, Shadid M, Romaine I, Dunlap N, Anklin C, Graves DE, Osheroff N (2008) Substituents on etoposide that interact with human topoisomerase IIα in the binary enzyme-drug complex: contributions to etoposide binding and activity. Biochemistry 46:4501–4509
Bentley R (2001) Bartolomeo Gosio, 1863–1944; an appreciation. Adv Appl Microbiol 48:229–250
Bevan MW, Franssen MCR (2006) Investing in green and white biotech. Nat Biotechnol 24:765–767
Birkinshaw JH, Raistrick H, Ross DJ (1952) Studies in the biochemistry of micro-organisms. 86. The molecular constitution of mycophenolic acid, a metabolic product of Penicillium brevi-compactum Dierckx. Part 3. Further observations on the structural formula for mycophenolic acid. Biochem J 50:630–634
Bode HB, Bethe B, Hofs R, Zeeck A (2002) Big effects from small changes: possible ways to explore nature’s chermical diversity. ChemBioChem 3:619–627
Bok JW, Hoffmeister D, Maggio-Hall LA, Murillo R, Glasner JD, Keller NP (2006) Genomic mining for Aspergillus natural products. Chem Biol 13:31–37
Brakage AA, Schroekh V (2011) Fungal secondary metabolites—strategies to activate silent genes. Fungal Genet Biol 48:15–22
Breinbauer R, Manger M, Scheck M, Waldmann H (2002) Natural product guided compound library development. Curr Med Chem 9:2129–2145
Breinbauer R, Vetter JR, Waldmann H (2002) From protein domains to drug candidates—natural products as guiding principles in the design and synthesis of compound libraries. Angew Chem Int Ed 41:2879–2890
Bringi V, Kadkade PG (1993) Enhanced production of taxol and taxanes by cell cultures of Taxus species. Patent WO93/17121
Bronson JJ, Barrett JF (2001) Quinolone, everninomycin, glycylcycline, carbapenem, lipopeptide and cephem antibiotics in clinical development. Curr Med Chem 8:1775–1793
Brown AG, Smale TC, King TJ, Hasenkamp R, Thompson RH (1976) Crystal and molecular structure of compactin, a new antifungal metabolite from Penicillium brevicompactum. J Chem Soc Perkins Trans I:1165–1170
Bull AT, Ward AC, Goodfellow M (2000) Search and discovery strategies for biotechnology: the paradigm shift. Microbiol Mol Biol Rev 64:573–606
Burrill GS (2002) Personalized medicine or blockbusterology. BioPharm 15(4):46–50
Busti E, Monciardini P, Cavaletti L, Bamonte R, Lazzarini A, Sosio M, Donadio S (2006) Antibiotic-producing ability by representatives of a newly discovered lineage of actinomycetes. Microbiology 152:675–683
Campbell WC (2012) History of avermectin and ivermectin, with notes on the history of other macrocyclic lactone antiparasitic agents. Curr Pharm Biotechnol 13:853–865
Cao Y, Langer R (2008) A review of Judah Folkman’s remarkable achievements in biomedicine. Proc Natl Acad Sci USA 105:13203–13205
Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS (2011) Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Translat Med 3(89):1–11
Cardenas ME, Shane Cutler N, Lorenz MC, Di Como CJ, Heitman J (1999) The TOR signaling cascade regulates gene expression in response to nutrients. Genes Devel 13:3271–3279
Carle P, Graeber M, Stuetz A (2000) Ascomycins: promising agents for the treatment of inflammatory skin diseases. Expert Opin Investig Drugs 9:69–77
Chan KY, Boucher ES, Gandhi PJ, Silva MA (2004) HMG-CoA reductase inhibitors for lowering elevated levels of C-reactive protein. Am J Health-Syst Pharm 61:1676–1681
Choi H-K, Kim S-I, Son J-S, Hong S–S, Lee H-S, Chung I-S, Lee H-J (2000) Intermittent maltose feeding enhances paclitaxel production in suspension culture of Taxus chinensis cells. Biotechnol Lett 2:1793–1796
Christoffersen RE (2006) Antibiotics. an investment worth making? Nat Biotechnol 24:1512–1514
Connors N, Pollard D (2004) Pneumocandin BO production by fermentation of the fungus Glarea lozoyensis: physiological and engineering factors affecting titer and structural analogue formation. In: An Z (ed) Handbook of Industrial Mycology. Marcel Dekker, NY, pp 515–538
Cragg GM, Newman DJ (2000) Antineoplastic agents from natural sources: achievements and future directions. Expert Opin Investig Drugs 9:2783–2797
Cruz MC, Del Poeta M, Wang P, Wenger R, Zenke G, Quesniaux VFJ, Movva NR, Perfect JR, Cardenas ME, Heitman J (2000) Immunosuppressive and nonimmunosuppressive cyclosporine analogs are toxic to the opportunistic fungal pathogen Cryptococcus neoformans via cyclophillin-dependent inhibition of calcineurin. Antimicrob Ag Chemother 44:143–149
Cruz MC, Goldstein AL, Blankenship J, Del Poeta M, Perfect JR, McCusker JH, Bennani YL, Cardenas ME, Heitman J (2001) Rapamycin and less immumosuppressive analogs are toxic to Candida albicans and Cryptococcus neoformans via FKBP12-dependent inhibition of TOR. Antimicrob Ag Chemother 45:3162–3170
Dejong JM, LiuY Bollon AP, Long RM, Jennewein S, Williams D, Croteau RB (2006) Genetic engineering of taxol biosynthetic genes in Saccharomyces cerevisiae. Biotechnol Bioeng 93:212–224
Demain AL (2002) Prescription for an ailing pharmaceutical industry. Nat Biotechnol 20:331
Demain AL (2006) From natural products discovery to commercialization: a success story. J Ind Microbiol Biotechnol 33:486–495
Demain AL, Adrio JL (2008) Contributions of microorganisms to industrial biology. Mol Biotechnol 38:41–55
Demain AL, Zhang L (2005) Natural products and drug discovery. In: Zhang L, Demain AL (eds) Natural products: drug discovery and therapeutic medicine. Humana Press, Totowa, pp 3–29
Deorukhkar A (2007) Back to basics: how natural products can provide the basis for new therapeutics. Expert Opin Investig Drugs 16:1753–1773
Dobson S, May T, Berriman M, Del Vecchio C, Fairlamb AH, Chakrabarti D, Bavic S (1999) Characterization of protein Ser/Thr phosphatases of the malaria parasite, Plasmodium falciparum: inhibition of the parasitic calcineurin by cyclophilin–cyclosporin complex. Molec Biochem Parasitol 99:167–181
Dunlap WC, Battershill CN, Liptrot CH, Cobb RE, Bourne DG, Jaspars M, Long PF, Newman DJ (2007) Biomedicinals from the phytosymbionts of marine invertebrates: a molecular approach. Methods 42:358–376
Dworkin M (2007) Lingering puzzles about myxobacteria. Microbe 2:18–24
Eckstein JW, Fung J (2003) A new class of cyclosporin analogues for the treatment of asthma. Expert Opin Investig Drugs 12:647–653
Einhorn LH (2002) Curing metastatic testicular cancer. Proc Natl Acad Sci (USA) 99:4592–4595
Endo A (2010) A historical perspective on the discovery of statins. Proc Jpn Acad Ser B 86:484–492
Endo A, Kuroda M, Tanzawa K (1976) Competitive inhibition of 3-hydroxyglutaryl coenzyme A reductase by ML-236A and ML-236B fungal metabolites, having hypocholesterolemic activity. FEBS Lett 72:323–326
Endo A (1980) Monocolin K, a new hypocholesterolemic agent that specifically inhibits 3-hydroxy-3-methylglutaryl coenzyme A reductase. J Antibiot 33:334–336
Engels EA, Biggar RJ, Hall HI, Cross H, Crutchfield A, Finch JL, Grigg R, Hylton T, Pawlish KS, McNeel TS, Goedert JJ (2008) Cancer risk in people infected with human immunodeficiency virus in the United States. Intl J Cancer 123:187–194
Ernst and Young (2000) The Ernst & Young Fifteenth Annual Report on the Biotechnology Industry. Ernst & Young LLP
Felnagle EA, Jackson EE, Chan YA, Podevels AM, Berti AD, McMahon MD, Thomas MG (2008) Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Molec Pharmaceut 5:191–211
Georgopapadakou NH (2001) Antifungals targeted to cell wall: focus on β-1,3-glucan synthase. Expert Opin Investig Drugs 10:269–280
Gerth K, Steinmetz H, Hofle G, Reichenbach H (2000) Studies on biosynthesis of epothilones: the biosynthetic origin of the carbon skeleton. J Antibiot 53:1373–1377
Gold BG (2000) Neuroimmunophilin ligands: evaluation of their therapeutic potential for the treatment of neurological disorders. Expert Opin Investig Drugs 9:2331–2342
Goodin S (2008) Novel cytotoxic agents: epothilones. Am J Health Syst Pharm 65(10 Suppl 3):S10–S15
Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, Jauch KW, Geissler EK (2002) Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nature Med 8:128–135
Guilder TAM, Moore BS (2009) Chasing the treasures of the sea—bacterial marine natural products. Curr Opin Microbiol 12:252–260
Hardt IH, Steinmetz H, Gerth K, Sasse F, Reichenbach H, Höfle G (2001) New natural epothilones from Sorangium cellulosum, Strains So ce90/B2 and So ce90/D13: isolation, structural elucidation, and SAR studies. J Nat Prods 64:847–856
Hopwood D, Malpartida F, Kieser HM, Ikeda H, Duncan J, Fujii I, Rudd BAM, Floss HG, Omura S (1985) Production of hybrid antibiotics by genetic engineering. Nature 314:642–644
Hranueli D, Cullum J, Basrak B, Goldstein P, Long PF (2005) Plasticity of the Streptomyces genome-evolution and engineering of new antibiotics. Curr Med Chem 12:1697–1704
Hugonnet J-E, Tremblay LW, Boshoff HI, Barry CE 3rd, Blanchard JS (2009) Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323:1215–1218
Jensen PR, Mincer TJ, Williams PG, Fenical W (2005) Marine actinomycete diversity and natural product discovery. Ant v Leeuwenhoek 87:43–48
Kingston DGI, Newman DJ (2002) Mother nature’s combinatorial libraries; their influence on the synthesis of drugs. Curr Opin Drug Disc Devel 5:304–316
Knowles J, Gromo G (2003) Target selection in drug discovery. Nat Rev 2:63–69
Kumaran RS, Muthumary JP, Hur B-K (2008) Taxol from Phyllosticta citricarpa, a leaf spot fungus of the angiosperm Citrus medica. J Biosci Bioeng 106:103–106
Lederberg J (2000) Pathways of discovery: infectious history. Science 288:287–293
Lee HJ, Lee HJ, Magesh V, Nam D, Lee EO, Ahn KS, Jung MH, Ahn KS, Kim DK, Kim JY, Kim SH (2008) Shikonin, acetylshikonin, and isobutyroylshikonin inhibit VEGF-induced angiogenesis and suppress tumor growth in Lewis lung carcinoma-bearing mice. Yakugaku Zasshi 128:1681–1688
Liu X-H, Zhao L-B, She Z-G, Lin YC (2004) Recent progress in bioactive metabolites of marine microorganisms. Chi J Antibiot 29:492–510
Lorence A, Nessler CL (2004) Camptothecin, over four decades of surprising findings. Phytochemistry 65:2735–2749
Manzoni M, Rollini M (2002) Biosynthesis and biotechnological production of statins by filamentous fungi and application of these cholesterol-lowering drugs. Appl Microbiol Biotechnol 58:555–564
Manzoni M, Bergomi S, Rollini M, Cavazzoni N (1999) Production of statins by filamentous fungi. Biotechnol Lett 21:253–257
Martinkova L, Juzlova P, Vesely D (1995) Biological activity of polyketide pigments produced by the fungus Monascus. J Appl Bacteriol 79:609–616
McAlpine J (1998) Unnatural natural products by genetic manipulation. In: DM Sapienza, LM Savage (Eds) Natural Products II: New Technologies to Increase Efficiency and Speed. Internat Bus Comm, Southboro, 251-278
Nathan C (2004) Antibiotics at the crossroads. Nature 431:899–902
Newman DJ, Cragg GM, Snader KM (2003) Natural products as sources of new drugs over the period 1981–2002. J Nat Prods 66:1022–1037
Newman DJ, Cragg GM (2005) in Natural Products: Drug Discovery and Therapeutic Medicine, L. Zhang and A.L. Demain, eds., pp. 129-168, Humana Press, Totowa, NJ
Newman DJ, Shapiro S (2008) Microbial prescreens for anticancer activity. SIM News 58:132–150
Omura S (2008) Ivermectin: 25 years old and still going strong. J Antimicrob Ag 31:91–98
Onyewu C, Blankenship JR, Del Poeta M, Heitman J (2003) Ergosterol biosynthesis inhibitors become fungicidal when combined with calcineurin inhibitors in Candida albicans, Candida glabrata, and Candida krusei. Antimicrob Ag Chemother 47:956–964
Overbye KM, Barrett J (2005) Antibiotics: where did we go wrong? Drug Disc Today 10:45–52
Pandey R, Chander R, Sainis KB (2007) Prodigiosins: a novel family of immunosuppressants with anti-cancer activity. Ind J Biochem Biophys 44:295–302
Park SR, Han AR, Ban Y-H, Yoo YJ, Kim EJ, Yoon YJ (2010) Genetic engineering of macrolide biosynthesis: past advances, current state and future prospects. Appl Microbiol Biotechnol 85:1227–1239
Park SR, Yoo YJ, Ban Y-H, Yoon YJ (2010) Biosynthesis of rapamycin and its regulation: past achievements and recent progress. J Antibiot 63:434–441
Patchett AA (2002) Alfred Burger award address in medicinal chemistry. Natural products and design: interrelated approaches in drug discovery. J Med Chem 45:5609–5616
Paululat T, Tang Y-Q, Grabley S, Thiericke R (1999) Combinatorial chemistry: the impact of natural products. Chim Oggi 17:52–56
Peng Y, Demain AL (1998) Properties of the hydroxylase in Actinomadura sp cells converting compactin to pravastatin. J Ind Microbiol Biotechnol 20:373–375
Piel J, Hui D, Wen G, Butzke D, Platzer M, Fusetani N, Matsunaga S (2004) Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc Natl Acad Sci USA 101:16222–16227
Pinter G, Batta G, Keki S, Mandi A, Komaromi I, Takacs-Novak K, Sztaricskai F, Roth E, Ostorhazi E, Rozgonyi F, Naesens L, Herczegh P (2009) Diazo transfer-click reaction route to new lipophilic teicoplanin and ristocetin aglycon derivatives with high antibacterial and anti-influenza virus activity: an aggregation and receptor binding study. J Med Chem 52:6053–6061
Pray L (2002) Strange bedfellows in transplant drug therapy. Scientist 16(7):36
Raskin I, Ribnicky DM, Komarnytsky S, Nabojsa I, Poulev A, Borisjuk N, Brinker A, Moreno DA, Ripoll C, Yakoby N, O’Neal JM, Cornwell T, Pastor I, Fridlender B (2002) Plants and human health in the twentieth century. Trends Biotechnol 20:522–531
Revill WP, Voda J, Reeves CR, Chung L, Schirmer A, Ashley G, Carney JR, Fardis M, Carreras CW, Zhou Y, Feng L, Tucker E, Robinson D, Gold BG (2002) Genetically engineered analogs of ascomycin for nerve regeneration. J Pharmacol Exp Ther 302:1278–1285
Rini B, Kar S, Kirkpatrick P (2007) Temsirolimus. Nature Rev/Drug Discov 6:599–600
Robert J, Jarry C (2003) Multidrug resistance reversal agents. J Med Chem 46:4805–4817
Rohde J, Heitman J, Cardenas ME (2001) The TOR kinases link nutrient sensing to cell growth. J Biol Chem 276:9583–9586
Rokem JS, Lantz AE, Nielsen J (2007) Systems biology of antibiotic production by microorganisms. Nat Prod Rep 24:1262–1287
Scheffler RJ, Colmer S, Tynan H, Demain AL, Gullo V (2013) Antimicrobials, drug discovery, and genome mining. Appl Microbiol Biotechnol 97:969–978
Schiewe H-J, Zeeck A (1999) Cineromycins: butyrolactones and ansamycins of the secondary metabolite pattern created by a single strain of Streptomyces. J Antibiot 52:635–642
Selitrennikoff CP (2001) Antifungal proteins. Appl Environ Microbiol 67:2883–2894
Serizawa N, Matsuoka T (1991) A two-component-type cytochrome P-450 monoxygenase system in prokaryotes that catalyzes hydroxylation of ML-236B to pravastatin, a tissue-selective inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Biochim Biophys Acta 1084:35–40
Smith L, Hillman JD (2008) Therapeutic potential of type A (I) lantibiotics, a group of cationic peptide antibiotics. Curr Opin Microbiol 11:401–408
Soetaert W, Vandamme EJ (2010) Industrial biotechnology. Sustainable growth and economic success. Wiley VCH, Weinheim
Song J-H (2008) What’s new on the antimicrobial horizon? Intl J Antimicrob Ag 32(Suppl 4):S207–S213
Stierle A, Strobel G, Stierle D (1993) Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science 260:214–216
Strobel GA (2002) Rainforest endophytes and bioactive products. Crit Rev Biotechnol 22:315–333
Strobel GA, Hess WM, Ford E, Sidhu RS, Yang X (1996) Taxol from fungal endophytes and the issue of biodiversity. J Indust Microbiol 17:417–423
Strohl WR (2004) Antimicrobials. In: Bull AT (ed) Microbial diversity and bioprospecting. ASM Press, Washington, DC, pp 336–355
Topliss JG, Clark AM, Ernst E, Hufford CD, Johnston GAR, Rimoldi JM, Weimann BJ (2002) Natural and synthetic substances related to human health. Pure Appl Chem 74:1957–1985
Udwary DW, Zeigler L, Asokar RN, Singan V, Lapidas A, Fenical W, Jensen PR, Moore BS (2007) Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinospora tropica. Proc Natl Acad Sci USA 104:10376–10381
Vaishnav P, Demain AL (2009) Industrial biotechnology (overview). In: Schaechter M (Ed) Encyclopedia of microbiology, third edition. Elsevier, 335–348
Vandamme EJ (2007) Microbial gems: microorganisms without frontiers. SIM-News 57:81–91
Veillard NR, Mach F (2002) Statins: the new aspirin? Cell Mol Life Sci 59:1771–1786
Vezina C, Kudelski A, Sehgal SN (1975) Rapamycin (AY-22989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot 28:721–726
Waldemann H, Breinbauer R (2002) Nature provides the answer. Screening 3(6):46–48
Wall ME, Wani MC (1996) Camptothecin and taxol: from discovery to clinic. J Ethnopharmacol 51:239–254
Wang JF, Li GL, Lu HY, Zhang ZH, Huang YJ, Su WJ (2000) Taxol from Tubercularia sp. strain TF5, an endophytic fungus of Taxus mairei. FEMS Microbiol Lett 193:249–253
Watkins KJ (2002) Fighting the clock. Chem Eng News 80(4):27–34
Wilkinson B, Micklefield J (2007) Mining and engineering natural-product biosynthetic pathways. Nature Chem Bio 3:379–386
Wong FT, Khosla C (2012) Combinatorial biosynthesis of polyketides—a perspective. Curr Opin Chem Biol 161:117–123
Wrigley SK (2004) Pharmacologically active agents of microbial origin. In: Bull AT (ed) Microbial diversity and bioprospecting. ASM Press, Washington, DC, pp 356–374
Xie X, Tang Y (2007) Efficient synthesis of simvastatin by use of whole-cell biocatalysis. Appl Environ Microbiol 73:2054–2060
Xie X, Wong WW, Tang Y (2007) Improving simvastatin bioconversion in Escherichia coli by deletion of bioH. Metab Eng 9:379–386
Xue Q, Ashley G, Hutchinson CR, Santi DV (1999) A multi-plasmid approach to preparing large libraries of polyketides. Proc Natl Acad Sci USA 96:11740–11745
Yang SS, Cragg GM, Newman DJ, Bader JP (2001) Natural product-based anti-HIV drug discovery and development facilitated by the NCI development therapeutics program. J Nat Prods 64:265–277
Yoneyama H, Katsumata R (2006) Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci Biotechnol Biochem 70:1060–1075
Youssef S, Stueve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, Zamvil SS (2002) The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 420:78–84
Zazopoulos E, Huang K, Staffa A, Liu W, Bachmann BO, Nonaka K, Ahlert J, Thorson JS, Shen B, Farnet CM (2003) A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nat Biotechnol 21:187–190
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Demain, A.L. Importance of microbial natural products and the need to revitalize their discovery. J Ind Microbiol Biotechnol 41, 185–201 (2014). https://doi.org/10.1007/s10295-013-1325-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-013-1325-z