
Abstract
In this article, we firstly report a highly alkali-tolerant fungal β-mannanase from Humicola insolens Y1. The full-length cDNA of the β-mannanase, designated as man5A, has an open reading frame of 1,233 bp that encodes a 411-amino acid polypeptide (Man5A) with a calculated molecular mass of 42.3 kDa. The deduced sequence of Man5A comprises a putative 20-residue signal peptide and a catalytic domain belonging to glycoside hydrolase family 5, and displays 61–85% identities with hypothetical proteins and 32–39% with experimentally verified fungal β-mannanases. Purified recombinant Man5A produced by Pichia pastoris has a specific activity of 1,122 U mg−1 and exhibits optimal activity at pH 5.5 and 70°C. Distinct from other reported fungal β-mannanases, Man5A is highly alkali tolerant, exhibiting 45 and 36% of the maximal activity at pH 8.0 and 9.0, respectively, and more than 10% activity even at pH 10.0. Moreover, Man5A has excellent pH stability at pH 5.0–12.0 and is highly thermostable at 50°C. The higher frequency of alkaline amino acids (Arg and Lys), greater pKa values of the catalytic residues, and more positively charged residues on the surface of Man5A might be the causes. Man5A has strong resistance to various neutral and alkaline proteases, retaining more than 97% of the activity after proteolytic treatment for 1 h. The superior characteristics of Man5A make it more advantageous for the application in the kraft pulp industry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mannans consist of a backbone of β-1,4-linked mannose or combination of glucose and mannose with side chains of α-1,6-linked galactose residues, and are the major constituents of the hemicellulose fraction in softwoods [13, 21]. For the efficient breakdown of mannans, catalytic action of several hydrolytic enzymes is necessary, including β-mannanase (EC 3.2.1.78), β-mannosidase (EC 3.2.1.25), α-galactosidase (EC 3.2.1.22), β-glucosidase (EC 3.2.1.21) and acetylmannan esterase (EC 3.1.1.6) [7, 17]. Among them, the crucial enzyme is β-mannanase that cleaves randomly the β-1,4-glycosidic linkages of the mannan backbone.
β-Mannanases are widely distributed in animals, plants, and microorganisms [6, 20, 27]. Among them, microbial β-mannanases are of considerable research and commercial importance because of their high activity and convenient isolation. Many β-mannanases have been purified and characterized from various microorganisms, including bacteria, fungi, and yeasts, and some β-mannanase genes have been cloned, sequenced, and expressed in heterologous systems [6, 17]. Based on the amino acid sequence and structural similarity among catalytic domains (http://www.cazy.org/), majority of β-mannanases are grouped into glycoside hydrolase (GH) families 5, 26, and 113 [9]. GH 5 contains majority of the β-mannanases that can be further divided into three subfamilies, GH 5–7, GH 5–8, and GH 5–10 [10, 38]. Bacterial β-mannanases are mainly classified into GH 5–8 with alkaline pH optima, and β-mannanases from eukaryota belong to GH 5–7 and 5–10 and act at acidic pHs [38]. Fungal β-mannanases are mainly confined into GH 5–7, and some of them can tolerate alkalinity up to pH 8.0 [6, 22, 32].
β-Mannanases are potential for applications in various industries. In the food industry, β-mannanases can be used for extraction of vegetable oils, clarification of juice, and production of soluble coffee [33]. Mannanases in animal feeds can result in decreased intestinal viscosity and improved weight gain and feed conversion efficiency [6, 33]. Moreover, mannanases are used in enzymatic bleaching of softwood pulps and laundry detergents [33, 34].
In this paper, we report the gene cloning and expression of a new β-mannanase that belongs to subfamily GH 5–7 from Humicola insolens Y1. Compared to other reported fungal β-mannanases, the purified recombinant enzyme showed higher catalytic activity and better stability under alkaline conditions.
Materials and methods
Strains and culture conditions
Humicola insolens Y1 was isolated from a forest soil sample of Hebei, China, and deposited in the Agricultural Culture Collection of China under registration number GCMCC 4573. The colony of strain Y1 was dark and short-tomentose. Conidiophores were dark and simply or rarely branched. Conidia were apical, one-celled, and globose or subglobose. Based on morphology and ITS sequence analysis [8], strain Y1 was identified to be H. insolens. Wheat bran medium was prepared by boiling 24 g wheat bran in 1 l of water for 15 min. The filtrate was adjusted to pH 6.0 and used to induce β-mannanase production in H. insolens Y1 at 42°C.
Escherichia coli trans 1 (Transgen, China) cultivated at 37°C in Luria–Bertani (LB) medium was used for gene cloning and sequencing. Pichia pastoris GS115 (Invitrogen, USA) cultivated at 30°C in yeast peptone dextrose (YPD) medium was used as the host for β-mannanase expression.
Vectors, media, and chemicals
The plasmids pGEM-T Easy (Promega, USA) and pPIC9 (Invitrogen) were used as the cloning and expression vectors, respectively.
Regeneration dextrose base (RDB) medium, minimal dextrose (MD) medium, minimal methanol (MM) medium, buffered glycerol complex (BMGY) medium, and buffered methanol complex (BMMY) medium were prepared according to the manual of Pichia Expression Kit (Invitrogen).
Locust bean gum (LBG) was purchased from Sigma (USA). The DNA purification kit, Genome Walking kit, and LA Taq DNA polymerase were purchased from TaKaRa (China). The Fungal DNA Mini kit from Omega Biotek (USA) and the RNeasy Plant Mini kit from Qiagen (Valencia, USA) were obtained, respectively. Restriction endonucleases and T4 DNA ligase were purchased from New England Biolabs (UK).
Cloning of the full-length chromosomal gene man5A
The DNA manipulations were carried out following the standard procedures [24]. The genomic DNA was extracted from the mycelia of H. insolens Y1 that grew in PDB medium using the Fungal DNA Mini kit. The core region of the β-mannanase gene was amplified by the degenerate primers specific for GH 5 β-mannanase genes [15] with the genomic DNA of strain Y1 as the template. The PCR product was ligated into the pGEM-T Easy vector and transformed into E. coli trans 1 cells for sequencing. The 5′ and 3′ flanking regions of the core region were obtained by thermal asymmetric interlaced (TAIL)-PCR [14] with the Genome Walking kit (TaKaRa). The amplified upstream and downstream products were sequenced and assembled with the core region to obtain the full-length gene.
Total RNA isolation and cloning of the cDNA fragment encoding the β-mannanase gene
Mycelia of H. insolens Y1 cultured in wheat bran medium for 3 days were collected and ground to a fine powder in liquid nitrogen. Total RNA was extracted with the RNeasy Plant Mini kit according to the manufacturer’s instructions. cDNA was synthesized in vitro from the mRNA template of H. insolens Y1 using the Reverse Transcription kit (Invitrogen). The full-length β-mannanase cDNA was obtained using the specific primers ManF (5′-ATGCACATCTCGACTGCAAGGTTGCTGACC-3′) and ManR (5′-CTAGTGAGCGTGACGGTTAAGTGCGTTCATC-3′) from total RNA by RT-PCR amplification.
Sequence analysis
BLASTx and PSI-BLAST [1] were used to compare a nucleotide query sequence translated in all reading frames and the putative translation products against the non-redundant protein database at the National Center for Biotechnology Information Web site (http://www.ncbi.nlm.nih.gov/Blast.cgi). The signal peptide was predicted by the SignalP 3.0 server [2]. Vector NTI 10.0 software was used to evaluate the identity and similarity of amino acid sequence of Man5A with other known proteins and to predict the molecular mass of the mature protein. Alignment of multiple protein sequences was accomplished using ClustalW software with the BLOSUM series as the protein weight matrix [30] and GeneDoc software [18].
A phylogenetic tree of Man5A and several characterized GH 5–7 β-mannanases was constructed based on their full-length amino acid sequences using the neighbor-joining algorithm in MEGA 4.1 [29]. The reliability was assessed by 1,000 bootstrap repetitions. A GH 26 β-mannanase was selected as the outgroup. Distance matrices for amino acids were calculated according to the PAM Matrix Model [5].
Homology modeling and surface charge analysis of Man5A were performed with Discovery Studio 2.5 MODELER (Accelrys, USA), using the β-mannanase from Trichoderma reesei (1QNO) as the template. The pKa values of the two catalytic glutamates and the amino acids that probably affect the pKa values were analyzed by PROPKA 3.0 (http://www.propka.ki.ku.dk/pka/).
Expression of man5A in P. pastoris
The gene fragment encoding the mature protein without signal peptide was amplified using primers man5AF-s: (5'-ACAGAATTCGCGCCGCATGTTCCCAAGACGTCGAAG-3', EcoRI site underlined) and man5AR: (5'-ACTGCGGCCGCCTAGTGAGCGTGACGGTTAAGTGCGTTCATCTC-3', NotI site underlined). The PCR product was digested with EcoRI and NotI and cloned in-frame at the downstream site of the α-factor signal peptide in pPIC9. The resulting plasmid, pPIC9-man5A, was linearized with BglII and then transformed into P. pastoris GS115 competent cells by electroporation (Bio-Rad, USA). Transformants were screened on RDB plates lacking histidine. The positive transformants with His+ phenotype were identified by PCR with using the 5′AOX and 3′AOX primers. The positive colonies were transferred to 5 ml of BMGY medium and grown at 30°C for 2 days. The cells were pelleted by centrifugation and resuspended in 1 ml BMMY containing 0.5% methanol for induction. The culture supernatant was collected for β-mannanase activity assay, and the transformant having the highest β-mannanase activity was used for subsequent analysis.
Purification of recombinant Man5A
Recombinant Man5A was purified from 3-day-old culture of induced P. pastoris. The culture supernatant was collected by centrifugation at 12,000×g for 10 min at 4°C to remove cell, concentrated using a vivaflow 50 ultrafiltration membrane with a 5-kDa molecular weight cut-off (Vivascience, Germany), and fractionated by ammonium sulfate (45–100% saturation). The fractions with β-mannanase activity were pooled and dissolved in 20 mM Tris–HCl (pH 8.0) and desalted by dialysis. The crude enzyme was loaded onto a HiTrap Q Sepharose XL 5 ml FPLC column (Amersham Pharmacia Biotech, Sweden) previously equilibrated with the same buffer. Proteins were eluted using a linear gradient of NaCl (0–0.3 M) in 20 mM Tris–HCl (pH 8.0). Fractions containing β-mannanase activity were concentrated by ultrafiltration as described above and further characterized.
SDS-PAGE, deglycosylation, and mass spectrometry analysis of Man5A
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) was carried out as described by Laemmli [11] with 12% polyacrylamide gel. The proteins were stained with Coomassie Brilliant Blue G-250. The protein concentration was determined by the Bradford method [4] using a protein assay kit (Bio-Rad). Purified recombinant β-mannanase was deglycosylated by Endo H according to the supplier’s instructions (New England Biolabs) and analyzed by SDS-PAGE. Liquid chromatography/electrospray ionization tandem mass spectrometry (LC–ESI–MS/MS) analysis of the purified protein was performed at the Institute of Zoology, Chinese Academy of Sciences.
Mannanase activity assay
β-Mannanase activity was determined by using the 3,5-dinitrosalicylic acid (DNS) method [16]. The reaction containing 100 μl of appropriately diluted enzyme sample and 900 μl of 0.5% (w/v) LBG in 0.1 M citric acid–Na2HPO4 (pH 5.5) was incubated at 70°C for 10 min, and terminated by the addition of 1.5 ml DNS reagent. The reaction was boiled in a water bath for 5 min, cooled to room temperature, and the absorbance at 540 nm was measured. The reaction system with the same enzyme sample added after DNS reagent was treated as a control. All experiments were run in triplicate. One unit of β-mannanase activity was defined as the amount of enzyme releasing 1 μmol of reducing sugar per min under the assay conditions (pH 5.5, 70°C, 10 min).
Effect of temperature, pH, and various chemicals on enzyme activity
The optimal pH and temperature, pH stability, thermal stability, and the effect of chemicals on the activity of purified recombinant Man5A were determined using the methods described previously [15].
The resistance of recombinant Man5A to different proteases, including trypsin, α-chymotrypsin, collagenase, subtilisin A, and proteinase K, was determined as described by Shi et al. [26]. After incubation of 30 or 60 min, the residual activity was measured under standard assay conditions (pH 5.5, 70°C, 10 min). The system without the presence of protease was used as a control.
Substrate specificity and kinetic parameters
The substrate specificity of recombinant purified Man5A was determined by measuring the enzyme activity after incubation of the enzyme in 0.1 M citric acid–Na2HPO4 (pH 5.5) containing 0.5% (w/v) of LBG, konjac flour, oat spelt xylan, or carboxymethyl cellulose–sodium (CMC–Na) or 5 mM p-nitrophenyl-β-d-mannopyranoside at 70°C for 10 min. The amount of released reducing sugars from LBG, konjac flour, oat spelt xylan, or CMC–Na was estimated as described above after addition of 1.5 ml DNS reagent. The amount of released p-nitrophenol was determined by measuring the absorbance at 420 nm after addition of 1.5 ml 1 M Na2CO3.
To determine the kinetic parameters (K m, app and V max, app), purified recombinant Man5A was incubated in 0.1 M citric acid–Na2HPO4 (pH 5.5) containing 0.5–10 mg ml−1 LBG at 70°C for 5 min. The data were calculated using the non-linear regression computer program GraFit. All assays had three independent experiments and every experiment included three samples.
Analysis of hydrolysis products
Reactions containing 50 U of purified Man5A and 500 μg of LBG or konjac flour in 500 μl citric acid–Na2HPO4 buffer (pH 5.5) were incubated at 70°C for 5 h. After hydrolysis, the enzyme was removed from the reaction using the Nanosep Centrifugal 3 K Device (Pall, USA). The hydrolysis products were analyzed using high-performance anion-exchange chromatography with a model 2500 system from Dionex (USA) [36]. Mannose, mannobiose, mannotriose, mannotetraose, and mannopentaose were used as standards.
Nucleotide sequence accession numbers
The nucleotide sequences of the H. insolens Y1 ITS regions and the β-mannanase gene (man5A) were deposited in the GenBank database under accession numbers HQ850154 and HQ850153, respectively.
Results
Cloning of β-mannanase gene and sequence analysis
A 227-bp DNA fragment was amplified from the genomic DNA of strain Y1 using degenerate primers M P1 and M P2 [15]. The amino acid sequence of this gene fragment showed 63% identity to the putative β-1,4-mannanase from Aspergillus terreus NIH2624. The 5′ and 3′ flanking regions of the core region were amplified by TAIL-PCR and assembled with the core region. The complete chromosomal gene of the β-mannanase consisted of 1,362 bp. The full-length cDNA sequence of man5A was 1,233 bp in length and encoded a polypeptide of 410 amino acids. Two introns, 178–258 and 589–636 bp, were identified within the coding sequence. SignalP analysis indicated the presence of a putative signal peptide at residues 1–20. The molecular mass and pI value of deduced Man5A were estimated to be 44.3 kDa and 6.4, respectively. Five potential N-glycosylation sites (Asn-Xaa-Thr/Ser-Zaa, where Zaa is not Pro) were found.
The BLAST search of the NCBI databases showed that deduced Man5A was most similar to a number of hypothetical proteins and putative β-mannanases. The most similar β-mannanases were the uncharacterized β-mannanase C from Aspergillus nidulans (68% identity) and those verified β-mannanases from Phanerochaete chrysosporium (39% identity), Aspergillus aculeatus (36% identity), and T. reesei (32% identity), all of which belong to GH 5–7.
Multiple amino acid sequence alignment of Man5A and 13 GH 5–7 β-mannanases with different pH optima was performed by ClustalW program (Fig. 1). Similar to all other GH 5 β-mannanases, Man5A contained two conserved catalytic glutamates (Glu 205 and Glu 322) and five active site residues (Arg 80, Asn 204, His 287, Tyr 289, and Trp 365) in close proximity to the catalytic center. Of all β-mannanases aligned, the conserved regions had only one variant residue, and six regions showed insertion–deletion polymorphism that might cause changes in the enzyme properties. Phylogenetic analysis of the same GH 5–7 β-mannanase sequences (Fig. 2) indicated that Man5A was closely related to the β-mannanases of Orpinomyces sp. PC-2, Armillariella tabescens and P. chrysosporium.

Multiple amino acid sequence alignment of Man5A from H. insolens Y1 (HQ850153.1) with 13 characterized GH 5–7 β-mannanases accomplished by the ClustalW software with the BLOSUM series as the protein weight matrix. GenBank accession number and pH optimum of each β-mannanase are presented. Similar and identical amino acids are indicated in grey and black, respectively. The two catalytic glutamate residues are indicated with an asterisk. Six regions showing insertion-deletion polymorphism are boxed

Phylogenetic dendrogram derived from the full-length amino acid sequences of Man5A from H. insolens Y1 (HQ850153.1) and 13 characterized GH 5–7 β-mannanases. The tree was constructed using neighbor-joining algorithm. GenBank accession number and microbial source of each β-mannanase are presented. The GH 26 β-mannanase from H. insolens (AAQ31840.1) is included as outgroup. Bootstrap values are expressed as percentages of 1,000 replications
The theoretical structure of Man5A was a classical (α/β)8 TIM-like-barrel fold. Compared to the homologous 1QNO that contained four disulfide bonds, Man5A had two external elongated loops on the surface that were located close to the conserved catalytic glutamates, and contained only one disulfide bond between Cys208 and Cys222.
Analysis of the model surface charge of Man5A at pH 7.4 using Discovery Studio 2.5 MODELER predicted that the surface near the substrate-binding cleft was mostly negative (Fig. 3a), and the opposite-side was positive (Fig. 3b). The pKa values of the catalytic residues of Man5A, 6.7 (Glu205) and 8.4 (Glu322), limit the pH range at which the enzyme can function [19]. Based on the analysis of PROPKA 3.0, several residues, Arg 207, Asp 151, Asp 268, Glu 252, and Glu 400, might affect the pKa values of the catalytic glutamates by forming hydrogen bonds and the coulombic interaction. The frequency of alkaline amino acids (Arg and Lys) was 11%, and most of them were located on the protein surface.

Charge distribution on the surface of a three-dimensional model of Man5A using Discovery Studio 2.5 MODELER. Charges on the surface were calculated at pH 7.4. Positive charges are depicted in black and negative charges in light grey. a The surface showing the substrate cleft with the active site. b The opposite-side surface
Expression, purification and deglycosylation of recombinant Man5A
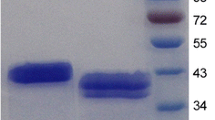
Pichia pastoris GS115 cells harboring pPIC9-man5A were induced with methanol to a final concentration of 0.5% for 72 h at 30°C. The highest β-mannanase activity was about 15 U ml−1. SDS-PAGE analysis of the crude enzyme showed a major protein band of about 60 kDa, which was higher than the predicted molecular weight (Fig. 4). The recombinant β-mannanase was purified to electrophoretic homogeneity by anion exchange chromatography. The specific activity of the purified recombinant Man5A was 1,122 U mg−1 after about 1.2-fold purification, with a final yield of 27.3%. In order to identify the purified protein, six peptides YKDSPVIAAWELANEPR, SPNCTPAVLSAWIAEMSAYIK, SLDRNHLVTWGGEGGFNR, TVEWTHQWIR, KPVVHEEYGWLTPDKR and NHNDGFTIYLDDEEAK obtained by LC–ESI–MS/MS were compared to the deduced amino acid sequence of Man5A. The complete matching indicated that the purified protein was indeed recombinant Man5A. The discrepancy between the actual molecular weight and that estimated based on amino acid sequence was probably due to glycosylation of the enzyme. After treatment with Endo H, purified recombinant Man5A had the apparent molecular mass of approximately 47 kDa (Fig. 4), still a little higher than the calculated molecular weight (44.3 kDa).

SDS-PAGE analysis of purified recombinant Man5A. Lanes: 1 standard protein molecular weight markers (LMW-SDS Marker, GE Healthcare, USA); 2 crude Man5A after membrane concentration and ammonium sulfate fraction; 3 purified recombinant Man5A; 4 purified recombinant Man5A after deglycosylation with Endo H
Effect of temperature and pH on enzyme activity and stability
LBG was used as the substrate for biochemical characterization of purified recombinant Man5A. The optimal pH for β-mannanase activity (at 70°C) was pH 5.5, and more than 75% of its maximal activity was retained within pH 5.5–7.0 (Fig. 5a). The enzyme was alkali tolerant, exhibiting over 35% activity at pH 9.0 and over 10% activity at pH 10.0, respectively. Recombinant Man5A was stable in the pH range of 5.0–12.0 (Fig. 5b). The optimal temperature was 70°C (at pH 5.5), and over 50% activity was retained in the temperature range of 50–80°C (Fig. 5c). The enzyme was stable at 50°C, and lost almost all of the activity after incubation at 60 and 65°C for 60 and 15 min, respectively (Fig. 5d).

Effects of pH and temperature on purified recombinant Man5A. a Effect of pH on β-mannanase activity. Activity was assayed at 70°C in buffers ranging from pH 4.0 to 11.0. b Effect of pH on the stability of β-mannanase activity. After incubation at 37°C for 1 h in buffers ranging from pH 3.0 to 12.0, the purified β-mannanase activity of recombinant Man5A was assayed in 0.1 M citric acid–Na2HPO4 (pH 5.5) at 70°C. c Effect of temperature on β-mannanase activity measured in 0.1 M citric acid–Na2HPO4 (pH 5.5). d Thermostability of Man5A. The enzyme was pre-incubated at 50, 60 or 65°C in 0.1 M citric acid–Na2HPO4 (pH 5.5), and aliquots were removed at specific time points to measure the residual activity at 70°C and pH 5.5. Each value represents the mean ± SD of triplicates
Effect of chemicals and proteases on the enzyme activity
The activity of purified recombinant Man5A was enhanced or not affected by Na+, K+, Li+, Mg2+, EDTA or β-mercaptoethanol. Partial inhibition (<50%) was observed in the presence of certain metal ions, including Ca2+, Cu2+, Ni2+, Zn2+, or 5 mM Cr3+. The enzyme was strongly inhibited by Pb2+, Hg2+, SDS, or 10 mM Cr3+ and Cu2+ (Table 1).
Purified recombinant Man5A was strongly resistant to all tested proteases. After treatment with trypsin, α-chymotrypsin, collagenase, subtilisin A, and proteinase K for 30 and 60 min, the recombinant enzyme retained more than 97% of the initial activity, respectively (data not shown).
Substrate specificity and kinetic parameters
Purified recombinant Man5A exhibited the highest activity towards LBG (100%) and lower activity on konjac flour (60.0%). No activity was detected against oat spelt xylan, CMC–Na or p-nitrophenyl-β-d-mannopyranoside.
The K m, app and V max, app values of Man5A using LBG as the substrate were 1.49 mg ml−1 and 1,122 μmol min−1 mg−1, respectively.
Analysis of hydrolysis product
Based on the HPAEC analysis, the hydrolysis products of LBG catalyzed by purified recombinant Man5A were 0.5% mannotriose, 23.1% mannotetraose, 5.7% mannopentaose, and 70.7% other mannan oligosaccharides. The composition of the hydrolysis products from konjac flour was 0.2% mannose, 10.1% mannobiose, 0.5% mannotriose, 15.4% mannotetraose, 3.0% mannopentaose, and 70.8% other oligosaccharides. The results indicated that Man5A was an endo-cleaving enzyme.
Discussion
A large number of β-mannanase genes of various sources have been identified in the rapidly expanding DNA sequence databases, and the properties of some of the β-mannanases have been evaluated. To the best of our knowledge, fungal β-mannanases generally function at acidic to neutral pH [32]. So far no alkaline fungal β-mannanases have been identified, and only a few fungal β-mannanases can tolerate alkalinity up to pH 8.0 [3, 25]. For example, T. reesei β-mannanase has an optimal pH at 5.0 and good stability at pH 2.5–7.0 [28]; P. chrysosporium β-mannanase has optimal activity at pH 4.0–6.0 and is stable from pH 4.0 to 8.0 [3]; β-mannanase from Orpinomyces sp. strain PC-2 was active from pH 3.8 to 8.0 [35]; the optimal pH for A. aculeatus β-mannanase is 5.0, and it is stable from pH 3.0 to 8.0 [25]. Compared with these close homologues, Man5A of H. insolens Y1 has a pH optimum at 5.5 and displays high activity at pH 9.0 and over 10% at pH 10.0. More over, it is stable up to pH 12.0. This is the first report of a fungal β-mannanase that has high activity and good stability at alkaline pH.
It has been reported that the pH dependence of protein stability and enzymatic activity is often determined by the pKa values of active-site residues [19, 31]. When enzymes perform at high pH values, their acid/base residues have to maintain higher pKa values [12]. The predicted pKa values of the catalytic glutamate residues of Man5A are 6.7 and 8.4, respectively, which are higher than that of most acid β-mannanases. Compared to the acid-active β-mannanases of T. reesei, P. chrysosporium and A. aculeatus that have 3.7–5.3% alkaline amino acids (Arg and Lys) [3, 23, 25], Man5A has more alkaline amino acid residues (11%), and most of the alkaline residues are located on the protein surface. In comparison to acid-stable β-mannanases [15, 23, 37], deduced Man5A has more positively charged residues on the surface. The higher pKa values, high frequency of alkaline amino acid residues and more positively charged residues might be the factors to influence the activity and stability of Man5A under neutral and alkaline conditions.
The optimal temperature of the recombinant Man5A is 70°C, similar to that of fungal β-mannanases [6]. However, the enzyme is stable only at 50°C. The higher optimal temperature than the stable temperature appears to be a common characteristic of fungal β-mannanases [3, 25, 28]. It is probably caused by the short assay time or the interaction between substrate and enzyme in the temperature optimum assay by comparison to the longer incubation time and no substrate present in the thermostability assay.
Compared with the β-mannanase from P. chrysosporium that has great application potential in the pulp and paper industry [3], Man5A has many superior properties, such as alkali tolerance, high stability under alkaline conditions, and strong resistance to proteases. These characteristics make Man5A more advantageous for the application in the kraft pulp industries. Moreover, Man5A had high activity at acid to neutral pH, and were potential for applications in the food and animal feed industries. Furthermore, study of this β-mannanase may contribute to a better understanding of the structure–function relationships of alkali-tolerant enzymes by comparisons of their properties with those of acid-active/stable enzymes from fungi.
References
Altschul SF, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bendtsen JD, Nielsen H, Heijne G, Brunak S (2004) Improved prediction of signal peptides: signalP 3.0. J Mol Biol 340:783–795
Benech RO, Li XM, Patton D, Powlowski J, Storms R, Bourbonnais R, Paice M, Tsang A (2007) Recombinant expression, characterization, and pulp prebleaching property of a Phanerochaete chrysosporium endo-β-1,4-mannanase. Enzyme Microbiol Technol 41:740–747
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Dayhoff MO, Schwartz RM, Orcutt BC (1978) A model of evolutionary change in proteins. In: Dayhoff MO (ed) Atlas of protein sequence and structure, vol 5, suppl 3. National Biomedical Research Foundation, Washington, pp 345–352
Dhawan S, Kaur J (2007) Microbial mannanases: an overview of production and applications. Crit Rev Biotechnol 27:197–216
Filho EXF (1998) Hemicellulases and biotechnology. In: Pandalai SG (ed) Recent research developments in microbiology. Research Signpost, Trivandrum, pp 165–176
Granchi L, Bosco M, Vicenzini M (1999) Rapid detection and quantification of yeast species during spontaneous wine fermentation by PCR-RFLP analysis of the rDNA ITS region. Appl Microbiol 87:949–956
Henrissat B, Bairoch A (1993) New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 293:781–788
Hilge M, Gloor SM, Rypniewski W, Sauer O, Heightman TD, Zimmermann W, Winterhalter K, Piontek K (1998) High-resolution native and complex structures of thermostable β-mannanase from Thermomonospora fusca—substrate specificity in glycosyl hydrolase family 5. Structure 6:1433–1444
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Le Nours J, Ryttersgaard C, Lo Leggio L, Østergaard PR, Borchert TV, Christensen LL, Larsen S (2003) Structure of two fungal β-1,4-galactanases: searching for the basis for temperature and pH optimum. Protein Sci 12:1195–1204
Liepman AH, Nairn CJ, Willats WGT, Sørensen I, Roberts AW, Keegstra K (2007) Functional genomic analysis supports conservation of function among cellulose synthase-like A gene family members and suggest diverse roles of mannans in plants. Plant Physiol 143:1881–1893
Liu YG, Whittier RF (1995) Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 25:674–681
Luo H, Wang Y, Wang H, Yang J, Yang Y, Huang H, Yang P, Bai Y, Shi P, Fan Y, Yao B (2009) A novel highly acidic β-mannanase from the acidophilic fungus Bispora sp. MEY-1: gene cloning and overexpression in Pichia pastoris. Appl Microbiol Biotechnol 82:453–461
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428
Moreira LRS, Filho EXF (2008) An overview of mannan structure and mannan-degrading enzyme systems. Appl Microbiol Biotechnol 79:165–178
Nicholas KB, Nicholas HB Jr (1997) GeneDoc: a tool for editing and annotating multiple sequence alignments. (Distributed by the author. http://www.psc.edu/biomed/genedoc).
Nielsen JE, McCammon JA (2003) Calculating pKa values in enzyme active sites. Protein Sci 12:1894–1901
Ootsuka S, Saga N, Suzuki KI, Inoue A, Ojima T (2006) Isolation and cloning of an endo-β-1,4-mannanase from Pacific abalone Haliotis discus hannai. J Biotechnol 125:269–280
Petkowicz CLO, Reicher F, Chanzy H, Taravel FR, Vuong R (2001) Linear mannan in the endosperm of Schizolobium amazonicum. Carbohydr Polym 44:107–112
Regalado C, Garcia-Almendarez BE, Venegas-Barrera LM, Tellez-Jurado A, Rodriguez-Serrano G, Huerta-Ochoa S, Whitaker JR (2000) Production, partial purification and properties of β-mannanases obtained by solid substrate fermentation of spent soluble coffee wastes and copra paste using Aspergillus oryzae and Aspergillus niger. J Sci Food Agric 80:1343–1350
Sabini E, Schubert H, Murshudov G, Wilson KS, Siika-Aho M, Penttilä M (2000) The three-dimensional structure of a Trichoderma reesei β-mannanase from glycoside hydrolase family 5. Acta Crystallogr D Biol Crystallogr 56:3–13
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Setati ME, Ademark P, van Zyl WH, Hahn-Hägerdal B, Stålbrand H (2001) Expression of the Aspergillus aculeatus endo-β-1,4-mannanase encoding gene (man1) in Saccharomyces cerevisiae and characterization of the recombinant enzyme. Protein Expr Purif 21:105–114
Shi P, Yuan T, Zhao J, Huang H, Luo H, Meng K, Wang Y, Yao B (2011) Genetic and biochemical characterization of a protease-resistant mesophilic β-mannanase from Streptomyces sp. S27. J Ind Microbiol Biotechnol 38:451–458
Shimahara H, Suzuki H, Sugiyama N, Nisizawa K (1975) Partial purification of β-mannanase from the konjac tubers and their substrate specificity in relation to the structure of konjac glucomannan. Agric Biol Chem 39:301–312
Stålbrand H, Siika-Aho M, Tenkanen M, Viikari L (1993) Purification and characterisation of two β-mannanases from Trichoderma reesei. J Biotechnol 29:229–242
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tynan-Connolly BM, Nielsen JE (2007) Redesigning protein pKa values. Protein Sci 16:239–249
van Zyl WH, Rosea SH, Trollopeb K, Görgensb JF (2010) Fungal β-mannanases: mannan hydrolysis, heterologous production and biotechnological applications. Process Biochem 45:1203–1213
Wong KKY, Saddler JN (1993) Applications of hemicellulases in the food, feed and pulp and paper industries. In: Coughlan MP, Hazlewood PG (eds) Hemicellulose and hemicellulases. Portland Press, London, pp 127–143
Wu G, Bryant MM, Voitle RA, Roland DA (2005) Effects of β-mannanase in corn-soy diets on commercial leghorns in second-cycle hens. Poultry Sci 84:894–897
Ximenes EA, Chen H, Kataeva IA, Cotta MA, Felix CR, Ljungdahl LG, Li XL (2005) A mannanase, ManA, of the polycentric anaerobic fungus Orpinomyces sp. strain PC-2 has carbohydrate binding and docking modules. Can J Microbiol 51:559–568
Yang P, Shi P, Wang Y, Bai Y, Meng K, Luo H, Yuan T, Yao B (2007) Cloning and overexpression of a Paenibacillus β-glucanase in Pichia pastoris: purification and characterization of the recombinant enzyme. J Microbiol Biotechnol 17:58–66
Zhao J, Shi P, Luo H, Yang P, Zhao H, Bai Y, Huang H, Wang H, Yao B (2010) An acidophilic and acid-stable β-mannanase from Phialophora sp. p13 with high mannan hydrolysis activity under simulated gastric conditions. J Agric Food Chem 58:3184–3190
Zhao Y, Zhang Y, Cao Y, Qi J, Mao L, Xue Y, Gao F, Peng H, Wang X, Gao GF, Ma Y (2011) Structural analysis of alkaline β-mannanase from alkaliphilic Bacillus sp. N16–5: implications for adaptation to alkaline conditions. PLoS One 6:1–12
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31172235) and the earmarked fund for China Modern Agriculture Research System (CARS-42) and the Key Program of Transgenic Plant Breeding (2009ZX08003-020B).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Luo, H., Wang, K., Huang, H. et al. Gene cloning, expression, and biochemical characterization of an alkali-tolerant β-mannanase from Humicola insolens Y1. J Ind Microbiol Biotechnol 39, 547–555 (2012). https://doi.org/10.1007/s10295-011-1067-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-011-1067-8