
Abstract
A novel fibrinolytic enzyme from Fusarium sp. CPCC 480097, named Fu-P, was purified to electrophoretic homogeneity using ammonium sulfate precipitation and ion exchange and gel filtration chromatography. Fu-P, a single protein had a molecular weight of 28 kDa, which was determined by SDS-PAGE and gel filtration chromatography. The isoelectric point of Fu-P determined by isoelectric focusing electrophoresis (IEF) was 8.1, and the optimum temperature and pH value were 45°C and 8.5, respectively. Fu-P cleaved the α-chain of fibrin (ogen) with high efficiency, and the β-chain and γ-γ (γ-)-chain with lower efficiency. Fu-P activity was inhibited by EDTA and PMSF, and the enzyme exhibited a high specificity for the chymotrypsin substrate S-2586. Fu-P was therefore identified as a chymotrypsin-like serine metalloprotease. The first 15 amino acids of the N-terminal sequence of Fu-P were Q-A-S–S-G-T-P-A-T-I-R-V-L-V–V and showed no homology with that of other known fibrinolytic enzymes. This protease may have potential applications in thrombolytic therapy and in thrombosis prevention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The incidence of thrombotic disorders including cerebral stroke, myocardial infarction, and venous thromboembolism are rapidly increasing worldwide, as is mortality due to these disorders [38]. During the past decade, thrombolytic agents have been extensively used in the therapeutic treatment of thrombosis [41]. Based on their different mechanisms of action, thrombolytic agents are divided into two types (1) plasminogen activators, such as tissue-type plasminogen activator (t-PA) [11] and urokinase [13]. This type of thrombolytic agent can activate the endogenous fibrinolytic system and generate plasmin from plasminogen. (2) Plasmin-like proteins, such as lumbrokinase [36] and various fibrolases [9], which can directly hydrolyze fibrin into fibrin degradation products, thereby dissolving the thrombi rapidly and completely. Despite their widespread use, these agents do have shortcomings such as excessive bleeding, low specificity to fibrin, and are relatively expensive [4, 18, 46]. Therefore, the search for less expensive and safer thrombolytic agents from various sources is still an urgent issue.
Effective thrombolytic agents have been identified and characterized from animals [6, 37, 49], plants [8, 35] and microorganisms [7, 16, 20]. Over the past few decades, thrombolytic agents from microorganisms have attracted more and more medical interest. These agents include NK produced by Bacillus natto [44], subtilisin DJ-4 [24] secreted by B. amyloliquefacien, subtilisin DFE [42] purified from B. amyloliquefacines, subtilisin CK [22] from Bacillus sp. strain CK-11-4 and streptokinase produced by Streptococcus hemolyticus [10]. Some fungi have also been found to have high fibrinolytic activity, for example, Aspergillus ochraceus 513 [2], Fusarium oxysporum [45], Fusarium sp. BLB [39], Rhizopus chinensis 12 [31], and P. chrysogenum H9 [14]. In addition, Kiminori Matsubara et al. [32–34] observed fibrinolytic enzymes from the marine algae Codium latum, C. divaricatum, and C. intricatum.
Endophytic fungi are a special and important group of microorganisms, which produce compounds with antibacterial and anti-tumor activities [15, 43], although reports on these compounds in thrombosis prevention are relative few. In this study, we found that the endophytic fungus Fusarium sp. CPCC 480097 isolated from chrysanthemum stems could produce a highly active fibrinolytic enzyme. We report on the purification and characterization of this fibrinolytic enzyme.
Materials and methods
Materials
Flour powder, Indian meal powder, corn powder and ground pea powder were purchased from the Beijing Comwin Pharm-Culture Co., Ltd. Human fibrinogen was purchased from the Green Cross China Biological Product Co., Ltd (Huainan, China). Human thrombin was purchased from the Shanghai Yingyue Biological Product Co., Ltd (Shanghai, China). Plasminogen, bovine serum albumin (BSA), phenylmethanesulfonyl fluoride (PMSF), tosyl-lysinechloromethylketone (TLCK), N-α-tosyl-phenylalanine chloromethyl ketone (TPCK), EDTA, Bz-Ile-Glu-(OR)-Gly-Arg-pNA (S-2222), H-d-Ile-Pro-Arg-pNA (S-2288), H-d-Phe-Pip-Arg-pNA (S-2238), H-d-Val-Leu-Lys-pNA (S-2251), pyroGlu-Gly-Arg-pNA (S-2444) and MeO-Suc-Arg-Pro-Tyr-pNA · HCl (S-2586) were obtained from Sigma (USA). Urokinase was obtained from the Suzhou Xinbao Pharmacia Co., Ltd (Suzhou, China). Sephadex G-25 desalting, MonoQ and Superdex 75 were products of Pharmacia Biotech (Uppsala, Sweden). Strain CPCC 480097 was held by Shanghai Health Creation Center of Biopharmaceutical R&D (Shanghai, China). Strain CPCC 480097 was deposited in CGMCC (ID. 2347) and the nucleotide sequences of internal transcriptional spacer (ITS) in the DDBJ/EMBL/GeneBank nucleotide sequence database under accession numbers: EU 363511. All other reagents were analytical grade and purchased commercially.
Culture conditions for enzyme production
The culture growth conditions of Fusarium sp. CPCC 480097 for enzyme production were preliminary optimized. The fungus strain of Fusarium sp. CPCC 480097 was initially cultured in 50 ml of liquid medium in a flask (500 ml) containing 1.5% Indian meal powder, 1.0% flour powder, 1.0% corn powder, 1.0% ground pea powder, 0.3% CaCO3, 0.1% MgSO4, 0.1% NaCl and 0.1% KH2PO4. The culture was incubated at 28°C on a rotary shaker at 220 rpm for 144 h. After the fermentation was completed, the culture broth was centrifuged for 30 min at 8,000 g. The supernatant and the microbial cells were harvested. The microbial cells were disrupted and the cell debris was removed by centrifugation. The fibrinolytic activity in the cells was about 10% as compared with that in the culture supernatant. Therefore, the supernatant was used for subsequent purification of the enzyme.
Purification of the fibrinolytic enzyme
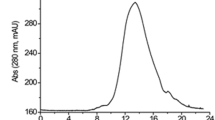
The culture supernatant was first treated using a two-step salting-out procedure at 4°C with ammonium sulfate, first at 40%, and then at 60%. The crude precipitate of the 60% solvent was collected by centrifugation at 8,000g for 30 min at 4°C. This precipitate was dissolved in a small volume of Tris–HCl buffer (20 mmol l−1, pH 7.4, buffer A) and then loaded onto a G-25 column (16 × 25 mm) equilibrated with buffer A for desalting. The protease-containing fraction was applied to a MonoQ column (10 × 100 mm) equilibrated with buffer A, and then eluted with a linear gradient of 0–1 mol l−1 NaCl in buffer A at a flow rate of 2 ml min−1. The active fraction was concentrated by lyophilized after desalting. The concentrated sample was dissolved in a small volume of buffer A and loaded onto a Superdex 75 column (16 × 600 mm) equilibrated with buffer A containing 50 mmol l−1 NaCl. The protein was eluted at a flow rate of 1 ml min−1 with buffer A. The active fraction was pooled, lyophilized, and analyzed for purity using SDS-PAGE. For all purification steps, the eluates were monitored by spectrophotometry at 280 nm. The activity of the protease was estimated with casein (DMC) as a substrate and by fibrin plate assay as described later.
Determination of protein concentration
Protein concentration was determined by the Bradford method [5], using BSA as a standard.
Determination of molecular weight and isoelectric point
The molecular weight of the enzyme was determined by SDS-PAGE and gel filtration chromatography on Superdex 75. SDS-PAGE was performed at room temperature by the method of Laemmli [29], using 12% polyacrylamide gel, and stained with Coomassie Brilliant Blue R250. Molecular weight was analyzed by running low molecular weight standard protein markers which comprised rabbit phosphorylase B (97.4 kDa), BSA (66.4 kDa), ovalbumin (44.2 kDa), carbonic anhydrase (29 kDa), trypsin inhibitor (20.1 kDa) and hen egg white lysozyme (14.3 kDa). Gel filtration chromatography was performed at room temperature, using a Superdex 75 column (AKTA FPLC) to estimate the molecular weight of the enzyme [30]. The elution buffer was 20 mmol l−1 Tris–HCl (pH 7.4) containing 50 mmol l−1 NaCl. A gel filtration protein marker comprised of glyceraldehydes (35 kDa), carbonic anhydrase (29 kDa), trypsinogen-PMSF (24 kDa), and trypsin-inhibitor (20.1 kDa) was used.
Isoelectric focusing electrophoresis (IEF) was performed using a Bio-Rad Model 111 Mini IEF Cell according to the manufacturer’s procedure. Broad isoelectric point (pI) calibration kits (proteins pI 4.45–9.60) were used as markers in the pI value determinations.
Determination of N-terminal amino acid sequence
The N-terminal amino acid sequence of the purified fibrinolytic enzyme was determined by the automated Edman method. Sequence alignment was analyzed with those of other known fibrinolytic enzymes.
Protease activity assay
Protease activity was determined with casein as a substrate according to the modified method of Kunitz [28]. The reaction was initiated by adding 20 μl of enzyme solution to 1 ml of 1% (w/v) casein in 20 mmol l−1 Tris–HCl buffer (pH 8.0). After 20 min at 37°C, the reaction was terminated by the addition of 3 ml of 5% (w/v) trichloroacetic acid (TCA), left for 20 min at room temperature, and centrifuged at 10,000g for 10 min at 4°C. The absorbance of the supernatant was measured at 280 nm. One unit (U) of protease activity was defined as the amount of enzyme that produced an increase in absorbance at 280 nm of 0.01 min−1 in the above conditions.
Fibrin-plate activity analysis
Fibrinolytic activity was determined by both the plasminogen-free fibrin plate method and the plasminogen-rich fibrin plate method with minor modifications [1]. Plasminogen-free fibrin plate was made up of a fibrinogen solution (5 ml of 0.5% human fibrinogen in 20 mmol l−1 Tris–HCl buffer, pH 7.4), 20 U of thrombin solution (same buffer), and 5 ml of 1% agarose in petri dishes (5 cm in diameter). The fibrin plate was heated at 80°C for 30 min to destroy other fibrinolytic factors. Plasminogen-rich fibrin plate containing 5 U plasminogen was not heated. To observe the fibrinolytic activity, 10 μl of enzyme solution was carefully dropped onto a fibrin plate and incubated at 37°C for 18 h. The activity of the fibrinolytic enzyme was estimated by measuring the dimension of the clear zone on the fibrin plate and plotting a calibration curve based on urokinase standard solutions. Three independent experiments were carried out to obtain average measurements of the zone diameters in these experiments.
Fibrinogenolytic activity analysis
Fibrinogenolytic activity was measured by the modified method of Koh [26]. In brief, 80 μl of 2% fibrinogen (prepared with 20 mmol l−1 Tris–HCl, pH 7.4) was incubated with 5 μg of the purified protease at 37°C. At various intervals a portion of the reaction solution was withdrawn and analyzed by SDS-PAGE.
Fibrinolytic activity analysis
Fibrin degradation analysis was performed as described by Datta et al. [12]. In brief, 20 μl of 2% human fibrinogen solution in 20 mmol l−1 Tris–HCl buffer, pH 7.4, was mixed with 20 μl of thrombin (5 U ml−1) dissolved in the same buffer. The fibrin clot was allowed to stand for 1 h at room temperature. Five microgram of the purified protease was placed on the clot surface and incubated at 37°C for various time intervals. The hydrolytic products from fibrin were analyzed by SDS-PAGE.
Effects of temperature and pH on enzyme fibrinolytic activity
The optimal temperature for the purified enzyme was determined by measuring residual activity in Tris–HCl buffer (pH 7.4) for 10 min at different temperatures (20–60°C). To measure its thermal stability, the purified protease was incubated in the same buffer for 4 h at 4–80°C. The remaining activity was determined using the casein method.
The optimal pH for the purified enzyme was determined by incubating the enzyme at room temperature in the following buffers, glycine–HCl (pH 2.0–6.0), Tris–HCl (pH 6.0–9.0), and glycine–NaOH (pH 9.0–11.5). All buffers were 20 mmol l−1. The effect of pH on enzyme stability was determined by incubating the enzyme for 4 h at 4°C in the above buffer. The remaining activity was determined using casein as a substrate.
Effects of metal ions and protease inhibitors on enzyme activity
The effects of metal ions on enzyme activity were investigated using MgCl2, ZnCl2, CoCl2, CaCl2, and CuSO4. The purified enzyme was pre-incubated in both the absence and presence of metal ions at various concentrations for 30 min at 37°C, and the protease activity was determined using casein as a substrate.
The effects of the protease inhibitors (EDTA, PMSF, TPCK, and TLCK) were also examined. The enzyme was pre-incubated with these protease inhibitors for 30 min at 37°C. After incubation, the effects were assessed with casein as a substrate.
Amidolytic activity of the enzyme
Amidolytic activities were measured spectrophotometrically, using chromogenic protease substrates such as S-2222 (for factor Xa), S-2288 (for t-PA), S-2238 (for thrombin), S-2251 (for plasmin and streptokinase-activated plasminogen), S-2444 (for urokinase), and S-2586 (for chymotrypsin). Activities were evaluated by mixing the purified enzyme [1 μg/200 μl of 20 mmol l−1 Tris–HCl (pH 7.4)] with 300 μl of each substrate (0.5 mmol l−1). After continuous measurement for 5 min at 37°C, the amount of p-nitroaniline released was determined by measuring the change in absorbance at 405 nm.
Results
Purification of the fibrinolytic enzyme
The fibrinolytic enzyme was purified using the steps listed in Table 1. Protease activity and fibrinolytic activity were used as indices of purification. After ammonium sulfate precipitation, a Mono-Q column was used to purify the enzyme, which yielded one major peak showing protease activity and fibrinolytic activity in the washed-out fraction. This result indicated that the enzyme did not bind to the Mono-Q and had a pI higher than 7.4. The washed-out fraction was further purified using a Superdex 75 column and eluted as four peaks. Only the third peak was noted to have protease and fibrinolytic activity, and showed a single band on SDS-PAGE (Fig. 1a). This active protein was named Fu-P. The enzyme was purified 158-fold, with a final yield of 6.8% after three purification steps as summarized in Table 1.

SDS-PAGE of purified Fu-P (a) and the mode of fibrinolytic action (b) and hydrolysis of fibrinogen by Fu-P (c). a Lane S a standard low molecular mass marker. Lane 1 recycled protein. b Analysis of fibrinolysis by Fu-P on plasminogen-rich fibrin plate (a) and plasminogen-free fibrin plate (b). 1 purified Fu-P (5 μg), 2 fermentation medium as a control, 3 20 mmol l−1 Tris–HCl buffer as a control. c a The hydrolytic products from fibrinogen were separated in 12% SDS-PAGE; b Lane 1, SDS-PAGE of fibrinogen incubated with inactivated Fu-P for 0 min; Lane 2 SDS-PAGE of fibrinogen incubated with inactivated Fu-P for 80 min. which showed that the fibrinogen alone was stable
Molecular weight and isoelectric point (pI)
The molecular weight of Fu-P was estimated to be 28 kDa by SDS-PAGE (Fig. 1a). This value was consistent with the molecular weight estimated by a Superdex 75 column, using AKTA FPLC (date not shown). Therefore, we concluded that the enzyme was a monomeric protein. The molecular weights of fibrinolytic enzymes from microbes have been reported to range from 18 to 52 kDa. The molecular weight of Fu-P was similar to those of the fibrinolytic enzyme from Fusarium sp. BLB [39], Nattokinase [16], subtilisin DFE [42], CK [22], subtilisin QK-2 [27], subtilisin DJ-4 [24], SW-1 [47] and subtilisin FS33 [48]. The pI of Fu-P was 8.1, as determined by IEF, which was comparable to that of subtilisin DEF [42], but lower than those of NK [16], subtilisin DJ-4 [24], SW-1 [47] and subtilisin FS33 [48] (Table 2).
The N-terminal amino acid sequence of the fibrinolytic enzyme
The N-terminal amino acid sequence of Fu-P was observed to be QASSGTPATIRVLVV. Multiple sequence alignment showed that Fu-P appeared to differ strongly from other reported fibrinolytic proteases (identity below 20%, Table 3). Thus, we strongly suggest that Fu-P is a novel protein.
Fibrinogenolytic and fibrinolytic activity
Fu-P formed a similar-sized clear lysis zone on both plasminogen-rich and plasminogen-free fibrin plates (Fig. 1b). The fibrinolytic activity measured by these two methods showed a difference of less than 2% difference, which indicated that Fu-P was not a plasminogen activator.
The fibrinogenolytic activity of Fu-P analyzed by SDS-PAGE showed that the α band disappeared first, followed by the β band, and then the γ chain (Fig. 1c). Similarly, when fibrin was incubated with Fu-P, the α band was rapidly hydrolyzed, and the β chain and γ–γ chain were slowly hydrolyzed (date not shown). This hydrolytic pattern was similar to that of other proteases purified from C. divaricatum and snake venom [3, 50], but was different from the fibrinolytic enzymes of the medicinal mushroom Cordyceps militaris [25] and B. subtilis FS33 [48].
Effects of pH and temperature on enzyme activity
The optimum pH of the purified protease was 8.5 (date not shown). Fu-P was stable over a pH range of 6–9 for 4 h at 4°C (date not shown). The optimum temperature of the enzyme was 45°C and the enzyme activity was stable below 37°C (date not shown).
Effects of metal ions and protease inhibitors on enzyme activity
The effects of protease inhibitors on the fibrinolytic activity of Fu-P were also examined and are shown in Table 3. Fu-P activity was strongly inhibited by the serine protease inhibitor PMSF and the metalloprotease inhibitor EDTA. However, TLCK had a weak inhibitory effect. These results indicated that Fu-P was a serine metalloprotease. Most known fibrinolytic enzymes from microbes are serine proteases and are not inhibited by EDTA. KA38 [23], SW-1 [47], an enzyme from R. chinensis 12 [31] and AMMP [30] are metalloproteases. However, the characteristics and the N-terminal amino acid sequence of Fu-P were different from those of these four enzymes.
The effects of metal ions on enzyme activity were also investigated (Table 4). The enzyme activity was found to be enhanced by Ca2+ (1 mmol l−1), Zn2+ (1 mmol l−1), but inhibited by Co2+ (3 mmol l−1), Cu2+, and Zn2+ (3 mmol l−1). Based on metal ion interactions with other fibrinolytic enzymes, all reported metallo fibrinolytic enzymes are inhibited by Cu2+ and Co2+. Some metallo fibrinolytic enzyme activities were enhanced by Ca2+ at 1 mM, such as the enzyme from the thermophilic fungus Oidiodendron flavum and AMMP. The fibrinolytic activity of KA38 was also enhanced by Zn2+ at 1 mM, but was decreased with increasing amounts of Zn2+. The mechanism of metal ions on fibrinolytic activity is not clear and requires further study.
Amidolytic activity of the enzyme
The amidolytic activity of Fu-P was measured using several chromogenic substrates (date not shown). The enzyme exhibited the highest degree of specificity for S-2586 (for chymotrypsin). According to the effects of inhibitors on Fu-P and its activities with chromogenic substrates, Fu-P can be classified as a chymotrypsin-like serine metalloprotease.
Discussion
Within the last few years, studies on fibrinolytic enzymes from microorganisms have attracted significant attention because of their potential use in thrombosis therapy. This article described the purification and characterization of a fibrinolytic enzyme (Fu-P) from Fusarium sp. CPCC 480097 isolated from chrysanthemum stems.
The overall findings of Fu-P in relation to the molecular weight, the isoelectric point, the N-terminal amino acid sequences, effect of inhibitors and metal ions, substrate specificity, and fibrino(geno)lytic activity indicate that Fu-P differs from other known fibrinolytic enzymes and is a novel fibrinolytic enzyme (Tables 2, 3).
Fu-P had strong fibrinolytic activity. The specific fibrinolytic activity was 76,111 U/mg. The units for the activity of fibrinolytic enzymes have been defined differently in many studies, making it difficult to compare absolute values. However, we noticed that the activity of Fu-P was much higher than that of subtilisin DJ-4 (4,664 U/mg), subtilisin QK-2 (41,000 U/mg), subtilisin FS33 (15,494.4 U/mg) and the enzyme from R. chinensis (2,143.4 U/mg), which were assayed by the same method. Fu-P also had a higher fibrinolytic activity than the typical fibrinolytic enzyme urokinase (Fig 2). The specific protease (caseinolytic) activity of the purified Fu-P was 40,920 U/mg. The ratio of fibrinolytic activity to caseinolytic activity was 1.86. This relative value was higher than that of other fibrinolytic enzymes, such as subtilisin DJ-4 (0.98) [24], CK (0.73) [22] and subtilisin Carlsberg (0.092) [22]. Therefore, Fu-P has relatively high substrate specificity for fibrin.

Comparison of the fibrinolytic activity of Fu-P and UK. The fibrinolytic activity was examined by fibrin plate assay. 1 UK (0.5 μg) 2 UK (1 μg) 3 UK (2 μg) 4 Fu-P (2 μg) 5 buffer (20 mmol l−1 Tris–HCl, pH 7.4) 6 distilled water. The calculated fibrinolytic activity of Fu-P was approximately 12% higher than that of UK according to the fibrin plate data
To investigate the mechanism of Fu-P induced fibrinolysis, the relative fibrinolytic activity of Fu-P on plasminogen-free and plasminogen-rich fibrin plates was examined. The results indicated that Fu-P acted via direct cleavage of fibrin and not by plasminogen activation like UK, SK and tPA. Therefore, secondary effects such as platelet activation related to plasmin formation could be avoided. This is a specific advantage of Fu-P over clinically used plasminogen activators. Fu-P also exhibited fibrinogenolytic activity by rapidly hydrolyzing the fibrinogen the α band, and then the β band and the γ band. Thus Fu-P could be termed a fibrinogenase-like enzyme. Fibrinogen, the final molecule in the coagulant cascade before the deposition of fibrin, is involved in primary haemostasis, platelet aggregation and is a major determinant of plasma viscosity [17]. A reduction in fibrinogen level decreases the incidence of thrombosis. From preliminary experiments with mice, we also found that Fu-P showed a significant in vivo inhibition of thrombus formation and in vitro degradation of blood clots (data not shown). Therefore, Fu-P may be used in thrombolytic therapy, and may also be used to prevent the formation of venous blood clots.
Taking account of these results, the fibrinolytic enzyme from Fusarium sp. CPCC 480097 may be a potential candidate for thrombolytic therapy or thrombosis prevention. Further investigation of this enzyme is underway.
References
Astrup T, Müllertz S (1952) The fibrin plate method for estimating fibrinolytic activity. Arch Biochem Biophys 40:346–351. doi:10.1016/0003-9861(52)90121-5
Batomunkueva BP, Egorov NS (2001) Isolation, purification and resolution of the extracellular proteinase complex of Aspergillus ochraceus 513 with fibrinolytic and anticoagulant activities. Microbiology 70:519–522. doi:10.1023/A:1012343718772
Bello CA, Hermogenes ALN, Magalhaes A (2006) Isolation and biochemical characterization of a fibrinolytic proteinase from Bothrops leucurus (white-tailed jararaca) snake venom. Biochimie 88:189–200. doi:10.1016/j.biochi.2005.07.008
Blann AD, Landray MJ, Lip GY (2002) An overview of antithrombotic therapy. BMJ 325:762–765. doi:10.1136/bmj.325.7367.762
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of proteins utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi:10.1016/0003-2697(76)90527-3
Cartwright T (1974) The plasminogen activator of vampire bat saliva. Blood 43:317–326
Chitte RR, Dey S (2000) Potent fibrinolytic enzyme from a thermophilic Streptomyces megasporus strain SD5. Lett Appl Microbiol 31:405–410. doi:10.1046/j.1365-2672.2000.00831.x
Chizhov AO, Dell A, Morris HR, Haslam SM, McDowell RA, Shashkov AS, Nifant’ev NE, Khatuntseva EA, Usov AI (1999) A study of fucoidan from the brown seaweed chorda filum. Carbohydr Res 20:108–119. doi:10.1016/S0008-6215(99)00148-2
Collen D, Lijene HR (1991) Basic and clinical aspects of fibrinolysis and thrombolysis. Blood 78:3114–3124
Collen D, Lijnen HR (1994) Staphylokinase, a fibrin-specific plasminogen activator with therapeutic potential. Blood 84:680–686
Collen D, Lijnen HR (2004) Tissue-type plasminogen activator: a histological perspective and personal account. J Thromb Haemost 2:541–546. doi:10.1111/j.1538-7933.2004.00645.x
Datta G, Dong A, Witt J, Tu AT (1995) Biochemical characterization of basilase, a fibrinolytic enzyme from Crotalus basiliscus. Arch Biochem Biophys 317:365–373. doi:10.1006/abbi.1995.1176
Duffy MJ (2002) Urokinase plasminogen activator and its inhibitor, PAI-I, as prognostic markers in breast cancer: From pilot to level 1 evidence studies. Clin Chem 48:1194–1197
EI-Aassar SA, EI-Badry HM, Abdel-Fattah AF (1990) The biosynthesis of proteases with fibrinolytic activity in immobilized cultures of Penicillium chrysogenum H9. Appl Microbiol Biotechnol 33:26–30
Filip P, Weber RWS, Sterner O, Anke T (2003) Hormonemate, a new cytotoxic and apoptosis-inducing compound from the endophytic fungus Hormonema dematioides. I. Identification of the producing strain, and isolation and biological properties of Hormonemate. Z Naturforsch 58:547–552
Fujita M, Nomura K, Hong K, Ito Y, Asada A, Nishimuro S (1993) Purification and characterization of a strong fibrinolytic enzyme (nattokinase) in vegetable cheese natto, a popular soybean fermented food in Japan. Biochem Biophys Res Commun 197:1340–1347. doi:10.1006/bbrc.1993.2624
Howard SC, Algra A, Rothwell PM (2008) Effect of age and glycaemic control on the association between fibrinogen and risk of acute coronary events after transient ischaemic attack or stroke. Cerebrovasc Dis 25:136–143. doi:10.1159/000112324
Hwang KJ, Choi KH, Kim MJ, Park CS, Cha J (2007) Purification and characterization of a new fibrinolytic enzyme of Bacillus licheniformis KJ-31, Isolated from Korean traditional Jeot-gal. J Microbiol Biotechnol 17:1469–1472
Jeong YK, Kim JH, Gal SW, Kim JE, Park SS, Chung KT, Kim YH, Kim BW, Joo WH (2004) Molecular cloning and characterization of the gene encoding a fibrinolytic enzyme from Bacillus subtilis strain AI. World J Microbiol Biotechnol 20:711–717. doi:10.1007/s11274-003-4514-5
Joeng YK, Park JU, Baek H, Park SH, Kong IS (2001) Purification and biochemical characterization of a fibrinolytic enzyme from Bacillus subtilis BK-17. World J Microbiol Biotechnol 17:89–92. doi:10.1023/A:1016685411809
Kato JY, Chi WJ, Ohnishi Y, Hong SK, Horinouchi S (2005) Transcriptional control by α-factor of two trypsin genes in Streptomyces griseus. J Bacteriol 187:286–295. doi:10.1128/JB.187.1.286-295.2005
Kim W, Choi K, Kim Y, Park H, Choi J, Lee Y, Oh H, Kwon I, Lee S (1996) Purification and characterization of a fibrinolytic enzyme produced from Bacillus sp. strain CK 11-4 screened from Chungkook-Jang. Appl Environ Microbiol 62:2482–2488
Kim HK, Kim GT, Kim DK, Choi WA, Park SH, Jeong YK, Kong IS (1997) Purification and characterization of a novel fibrinolytic enzyme from Bacillus sp. KA38 originated from fermented fish. J Ferment Bioeng 84:307–312. doi:10.1016/S0922-338X(97)89249-5
Kim SH, Chio NS (2000) Purification and characterization of subtilisin DJ-4 secreted by Bacillus sp. strain DJ-4 screened from Doen-Jang. Biosci Biotechnol Biochem 64:1722–1725. doi:10.1271/bbb.64.1722
Kim JS, Sapkota K, Park SE, Choi BS, Kim S, Hiep NT, Kim CS, Choi HS, Kim MK, Chun HS, Park Y, Kim SJ (2006) A fibrinolytic enzyme from the medicine mushroom Cordyceps militaris. J Microbiol 44:622–631
Koh YS, Chung KH, Kim DS (2001) Biochemical characterization of a thrombin-like enzyme from snake. Toxcion 39:555–560. doi:10.1016/S0041-0101(00)00169-0
Ko JH, Yan JP, Zhu L, Qi YP (2004) Identification of two novel fibrinolytic enzymes from Bacillus subtilis QK02. Comp Biochem Physiol C Toxicol Pharmacol 137:65–74. doi:10.1016/j.cca.2003.11.008
Kunitz M (1974) Crystalline soybean trypsin inhibitor. J Gen Physiol 30:291–310. doi:10.1085/jgp.30.4.291
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi:10.1038/227680a0
Lee SK, Kim JS, Kim JE, Sapkota K, Shen MH, Kim S, Chun HS, Yoo JC, Choi HS, Kim MK, Kim SJ (2005) Purification and characterization of fibrinolytic enzyme from cultured mycelia of Armillaria mellea. Protein Expr Purif 43:10–17. doi:10.1016/j.pep.2005.05.004
Liu XL, Du LX, Lu FP, Zheng XQ, Xiao J (2005) Purification and characterization of a novel fibrinolytic enzyme from Rhizopus chinensis12. Appl Microbiol Biotechnol 67:209–214. doi:10.1007/s00253-004-1846-5
Matsubara K, Sumi H, Hori K, Miyazawa K (1998) Purification and characterization of two fibrinolytic enzymes from a marine green alga, Codium intricatum. Comp Biochem Physiol Biochem Mol Biol 119:177–181. doi:10.1016/S0305-0491(97)00303-9
Matsubara K, Hori K, Matsuura Y, Miyazawa K (1999) A fibrinolytic enzyme from a marine green alga, Codium latum. Phytochemistry 52:993–999. doi:10.1016/S0031-9422(99)00356-8
Matsubara K, Hori K, Matsuura Y, Miyazawa K (2000) Purification and characterization of a fibrinolytic enzyme and identification of fibrinogen clotting enzyme in a marine green alga, Codium divaricatum. Comp Biochem Physiol Biochem Mol Biol 125:137–143. doi:10.1016/S0305-0491(99)00161-3
Maurer HR (2001) Bromelain: biochemistry, pharmacology and medical use. Cell Mol Life Sci 58:1234–1245. doi:10.1007/PL00000936
Mihara H, Sumi H, Yoneta T, Mizumoto H, Ikeda R, Seiki M, Maruyama M (1991) A novel fibrinolytic enzyme extracted from the earthworm, Lumbricus rubellus. Jpn J Physiol 41:461–472. doi:10.2170/jjphysiol.41.461
Mihara H (1983) Fibrinolytic enzyme extracted from earthworm. Thromb Haemost 50:258–263
Mine Y, Wong AHK, Jiang B (2005) Fibrinolytic enzymes in Asian traditional fermented foods. Food Rev Int 38:243–250. doi:10.1016/j.foodres.2004.04.008
Mitsuhiro U, Toshihiro K, Kazutaka M, Takumi N (2007) Purification and characterization of fibrinolytic alkaline protease from Fusarium sp. BLB. Appl Microbiol Biotechnol 74:331–338. doi:10.1007/s00253-006-0621-1
Nagwa A, Tharwat H (2006) Purification and biochemical characterization of fibrinolytic enzyme produced by thermophilic fungus Oidiodendron flavum. Biotechnology 5:160–165
Ouriel K (2002) Current status of thrombolysis for peripheral arterial occlusive disease. Ann Vasc Surg 16:797–840. doi:10.1007/s10016-001-0318-y
Peng Y, Huang Q, Zhang RH, Zhang Y (2003) Purification and characterization of a fibrinolytic enzyme produced by Bacillus amyloliquefaciens DC-4 screened from douchi, a traditional Chinese soybean food. Comp Biochem Physiol Biochem Mol Biol 134:45–52. doi:10.1016/S1096-4959(02)00183-5
Strobel GA (2003) Endophytes as sources of bioactive products microbes and infection. Microbes Infect 5:535–544. doi:10.1016/S1286-4579(03)00073-X
Sumi H, Hamada H, Tsushuma H, Mihara H, Muraki H (1987) A novel fibrinolytic enzyme (nattokinase) in the vegetable cheese Natto, a typical and popular soybean food in the Japanese diet. Experientia 43:1110–1111. doi:10.1007/BF01956052
Tao S, Peng L, Lu BH, Liu DM, Liu ZH (1998) Successive cultivation of Fusarium oxysporum on rice chaff for economic production of fibrinolytic enzyme. Bioprocess Eng 18:379–381
Turpie AG, Chin BS, Lip GY (2002) Venous thromboembolism: treatment strategies. BMJ 325:947–950. doi:10.1136/bmj.325.7370.947
Wang J, Wang M, Wang Y (1999) Purification and characterization of a novel fibrinolytic enzyme from Streptomyces sp. Chin J Biotechnol 15:83–89
Wang CT, Ji BP, Li B, Rob N, Li PL, Ji H, Chen LF (2006) Purification and characterization of a fibrinolytic enzyme of Bacillus subtilis DC33, isolated from Chinese traditional Douchi. J Ind Microbiol Biotechnol 33:750–758. doi:10.1007/s10295-006-0111-6
Zhang Y, Wisner A, Xiong YL, Bon C (1995) A novel plasminogen activator from snake venom. J Biol Chem 270:10246–10255. doi:10.1074/jbc.270.17.10246
Zhang YL, Cui JY, Zhang R, Wang YP, Hong M (2007) A novel fibrinolytic serine protease from the polychaete Nereis (Neanthes) virens (Sars): Purification and characterization. Biochimie 89:93–103. doi:10.1016/j.biochi.2006.07.023
Acknowledgments
This work was support by the National Infrastructure of National Resources for Science and Technology (grant no 2005DKA21203). Isoelectric point and the N-terminal amino acid sequence of Fu-P were measured by Shanghai Institutes for Biological Sciences (Chinese Academy of Sciences).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wu, B., Wu, L., Chen, D. et al. Purification and characterization of a novel fibrinolytic protease from Fusarium sp. CPCC 480097. J Ind Microbiol Biotechnol 36, 451–459 (2009). https://doi.org/10.1007/s10295-008-0516-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-008-0516-5