
Abstract
The fed-batch process using glucose as the sole source of carbon and energy with exponential feeding rate was carried out for high cell density cultivation of recombinant Escherichia coli BL21 (DE3) expressing human granulocyte-colony stimulating factor (hG-CSF). IPTG was used to induce the expression of hG-CSF at 48 g dry cell wt l−1 during high cell density culture of recombinant E. coli BL21 (DE3) [pET23a-g-csf]. The final cell density, specific yield and overall productivity of hG-CSF were obtained as ~64 g dry cell wt l−1, 223 mg hG-CSF g−1 dry cell wt and 775 mg hG-CSF l−1 h−1, respectively. The resulting purification process used cell lysis, inclusion body (IB) preparation, refolding, DEAE and Butyl-Sepharose. Effects of different process conditions such as cell lysis and washing of IB were evaluated. The results reveal that the cells lyzed at 1,200 bar, 99.9% and Triton removed about 64% of the LPS but sarcosyl had no effect on removal of nucleic acids and LPS. Further analysis show that DEAE column removes DNA about 84%. Cupper concentration was identified as parameter that could have a significant impact on aggregation, as an unacceptable pharmaceutical form that decrease process yields. The purity of purified hG-CSF was more than 99%. Also the comparison of activity between purified hG-CSF and commercial form do not show valuable decrease in activity in purified form.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Escherichia coli is the most commonly used host for heterologous protein production because it is a well-characterized organism in the genetics, physiology and cultivation condition [1–3]. Growing E. coli to high density is currently the method of choice for the production of recombinant proteins, mainly because of the high volumetric productivity associated with this method. Moreover, high cell density cultivation (HCDC) techniques have several additional advantages such as: reduced culture volume, enhanced downstream processing, reduced fermentation effluents, lower production costs and reduced investment in equipment [1, 2].
Granulocyte colony-stimulating factor (G-CSF) is a 19.6 kDa glycoprotein consisting of 174 amino acid residues, which is produced by activated macrophages, endothelial cells and fibroblasts. Injecting pharmacological doses of G-CSF into normal mice results in a marked increase in the number of granulocytes and their precursors without significant hematopoietic effects on other lineages of blood cells. These studies indicate that G-CSF is a potent and highly specific stimulator of neutrophil production. From the viewpoint of target cell specificity, G-CSF is an attractive vector for delivering biological substances into normal or abnormal granulocytes and their precursors [4, 5].
The advances in recombinant DNA technology have facilitated the production of biological medicines and recombinant human G-CSF (rhG-CSF) is now available for clinical use in two forms: non-glycosylated and glycosylated. The non-glycosylated form of hydrophobic protein, known as filgrastim, contains an extra methionine at its N-terminus, consisting of 175 aminoacids, derived from expression in E. coli [6].
In order to produce recombinant proteins in E. coli with high yield, over-expression of the recombinant protein in a fermentation process and a purification procedure allowing efficient recovery of the protein from the resultant biomass are necessary. Several methods based on multiple chromatographic steps have been reported for the purification of rhG-CSF [7–9].
Earlier reports indicate that the levels of rhG-CSF expressed in E. coli was at moderate to high level [10–13], the yield of final product was very poor and far from satisfactory. This is probably due to inefficient downstream process technologies like isolation of protein inclusion body (IB) with low purity and recovery, mis folding, aggregate formation and unoptimized conditions of protein refolding and chromatographic processes. Such multi-step procedures are cumbersome and the overall yields are low. For example, a purification scheme involving cell lysis following denaturation, Talon resin and on column renaturation was described for purification of His-tagged G-CSF. By this method, it was possible to isolate approximately 0.5–2 mg of refolded G-CSF per liter of growth medium [13]. Kuga et al. [7] described a procedure involving IB solubilization, refolding by dialysis and DEAE-Sepharose chromatography. A purification process based on size-exclusion chromatography (Sephacryl S-200), renaturation and CM-Sepharose with overall recovery of about 20% was reported by Wingfield et al. [8].
In this research, we have focused on the production of hG-CSF from recombinant E. coli BL21 (DE3) which over-expressed hG-CSF in the form of insoluble inclusion bodies. The fed-batch culture was performed and optimized to maximize overall productivity of hG-CSF through HCDC. Also, our work describes a process development of purification method of hG-CSF and describes the procedures and results of process development studies that were performed for the purification strategy.
Materials and methods
Microorganism
The E. coli strain BL21 (DE3) (Novagen, Inc.) was used as the host for hG-CSF expression. Transformation of this strain was performed using commercially available plasmid, pET23a inducible expression vector (Novagen, Inc.), in which g-csf gene was inserted into the NdeI and EcoRI sites [14].
Media and inoculum preparation
LB-agar medium was used for cultivation of E. coli strain at the plate and a defined medium (M9 modified medium) used for preparation of seed culture and batch fermentation [15].
Stock solutions of glucose and MgSO4 were sterilized separately for 30 min at 121 °C and mixed aseptically to make the feeding solutions.
In order to cultivation the recombinant E. coli, it was removed from −70 °C freezer and grown at 37 °C for 24 h on LB plates supplemented with ampicillin (100 mg l−1). One colony from LB plate was transferred into 100 ml defined medium (pH = 7) supplemented with ampicillin (100 mg l−1) in the 500-ml flask and incubated overnight at 37 °C and 200 rpm on a rotary shaker incubator (Kühner, Switzerland).
Bioreactor system and fed-batch fermentation
The fed-batch fermentation was carried out in a 2-l bench top bioreactor (INFORS AG, Switzerland). The initial batch culture was started by inoculation of 100 ml of overnight-incubated seed culture (0.4–0.6 g l−1 of dry cell mass) to 1,000 ml of defined medium supplemented with ampicillin (100 mg l−1) in the bioreactor. Cultivation condition was controlled at 37 ± 0.5 °C, airflow rate of 1 vvm, pH 7 ± 0.05, and 400-rpm. Foam formation was suppressed by the manually addition of silicon-antifoaming reagent when necessary. Dissolved oxygen was measured using a polarographic electrode (Ingold, Mettler Toledo, Germany) and controlled at 20–30% of air saturation by controlling of both air flow and stirrer speed. During the fed-batch operation, pure oxygen was used for enrichment of entering air. The pH was adjusted at 7.0 ± 0.05 by addition of 25(w/w)% NH4OH (or 2 M NaOH) or 1 M H3PO4 while ammonium was maintained between 0.1 and 1.5 g l−1.
After depletion of the initial carbon source (glucose) in the batch medium, as indicated by a rapidly increase in the dissolved oxygen concentration, the feeding was initiated and the flow rate was increased stepwise based on exponential feeding strategy. The exponential feeding rate was determined by a simple mass balance equation of the cell and substrate [16].
where V is the medium volume in the bioreactor (l), X is the biomass concentration in the bioreactor (g dry cell wt l−1), t is the time (h), μ is the specific growth rate (h−1), S is the glucose concentration in the bioreactor (g l−1), S 0 is the glucose concentration in the feeding solution (g l−1), F is the feeding rate (l h−1), and Y x/s is the glucose yield coefficient (g dry cell weight g−1 glucose).
Assuming a quasi-steady state exists for the substrate concentration, (d/dt) (VS) = 0, and constant volume fed-batch fermentation. If assume Y x/s is constant, then by integrating and substituting Eq. (1) into (2), Eq. (3) will be [15]:
where X 0 (g dry cell wt l−1) is the biomass concentration when the feeding is started, V 0 (l) is the medium volume in the bioreactor at the start of feeding, M s (t) is the mass flow rate of glucose(g glucose h−1).
Cell disruption and inclusion bodies preparation
The cells were harvested by centrifugation (6K-15, Sigma) at 5,000 rpm for 5 min at 4 °C. Pellet (1.5–2.0 g) was suspended in 1:20 w/v of buffer a1 (Tris–HCl, 20 mM pH 8.0, 2 mM EDTA, 0.002% PMSF) by mechanical homogenizer (Diax-100 Heidolph) at 4 °C and cells disrupted by twice passing through a high pressure homogenizer (NS1001l-PANDA2K, Niro Soavi S.p.A) at 800 and 1,200 bar and suspension of lysed cells centrifuged at 10,000 rpm for 20 min at 4 °C. The pellet was resuspended in 1:20 w/v of buffers, a2 (Tris–HCl, pH 8.0, 2 mM EDTA, 0.002% PMSF, and 1% Triton X-100) using a mechanical homogenizer and centrifuged at 10,000 rpm for 20 min at 4 °C. The pellet obtained from previous step was resuspended again in buffer a2 using mechanical homogenizer and centrifuged at 10,000 rpm for 20 min at 4 °C. The pellet was resuspended in buffer a3 (Tris–HCl, 20 mM pH 8.0, 2 mM EDTA, 0.002% PMSF, 1% sarcosyl) and was centrifuged similar to protocol described for buffer a2.
Solubilization of inclusion bodies
The washed pellet, containing purified inclusion bodies, was solubilized in 1:40 w/v of buffer b (Tris–HCl buffer, 20 mM, pH 8.0, 8 M urea, 2 mM EDTA, 0.002% PMSF) at 4 °C. The protein was then reduced by 0.1 mM dithiothreitol for 5 h at 4 °C.
Refolding of hG-CSF
After solubilization, the solution was centrifuged in 11,500 rpm for 30 min at 4 °C. The solubilized protein was refolded with step–step dialysis method against decreasing concentration of urea (8–0 M) in 20 mM, Tris pH 8.0 containing 5, 20, 40 and 60 μM CuSO4. After refolding, the solution was centrifuged at 10,000 rpm, 4 °C, for 10 min, and the soluble aggregated form of protein was measured by A 340 nm [17].
The chromatography procedures
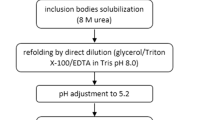
Experiments were performed using 1.6 × 10 and 2.5 × 10 cm columns (Amersham Pharmacia Biotech). Schemes 1, 2, 3 illustrate the cell lysis, IB solubilization, DEAE and Butyl column dimensions, buffers, procedures and other operating conditions for purification of hG-CSF. Two columns were used and the obtained fractions were analyzed by a variety of analytical tools that are described in the analytical methods section.

Operating procedure for cell lysis, IB washing and refolding

Operating procedure for DEAE column

Operating procedure for Butyl column
Analytical methods
The optical density (OD) was measured at 600 nm and converted to the dry cell weight by an appropriate calibration curve [18]. Dry cell weight was determined with a 5-ml culture sample, which was centrifuged at 5,000 rpm for 5 min, the pellet was collected and washed twice with de-ionized water and dried at 105 °C to constant weight [18]. Glucose and ammonia analyzed by enzymatic kits (ChemEnzyme Co., I.R. Iran) according the procedure suggested by the supplier. Enzymatic kit (Boehringer Mannheim/R-Biopharm, Germany) was used to measure acetate according the procedure suggested by the supplier. Expression level of recombinant hG-CSF and purity during process were determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 12.5% (w/v) polyacrylamide gels stained with 0.1% (w/v) Coomassie Brillient Blue R250 and quantified by densitometry.
The stability of plasmid in the recombinant E. coli strain was measured by aseptically sampling of the bioreactor at different dry cell weight. The sample was diluted with sterile solution of NaCl (9 g l−1) to yield 100–300 colonies per plate on LB-agar medium and incubated at 37 °C for 16 h. All colonies on three plates were transferred on selective LB-agar plates (supplemented with 100 mg l−1 of ampicillin) by replica plating method. Plasmid stability was calculated by taking the ratio of the average number of colonies from three selective LB-agar plates to the average from three non-selective LB-agar plates [19].
Colony counting was used as a quantitative method to measure the cell lysis efficiency. LAL kit was used as a quantitative assay for removal of bacterial endotoxins. The ELISA polyclonal antibody kit was designed for measurement of expressed hG-CSF and A 260 was used for measurement of removal of nucleic acid during this process. Protein concentration was determined according to Bradford assay [20]. Reducing SDS-PAGE was carried out according to Laemmli method [21], upper gel 4% and separating gel 12.5%. The purity of hG-CSF (%) in the gel fractions was determined by scanning of Coomassie brilliant R250 stained gels using a densitometer. Biological assay was performed by NFS60 proliferation assay. NFS60 cells were maintained in IMDM (GIBCO-BRL) with 15% fetal calf serum (FCS) and 5 ng ml−1 recombinant hG-CSF [22]. Prior to assays, exponentially growing cells were washed free of G-CSF and resuspended in IMDM with 5% FCS. Aliquots of 5 × 104 cells were incubated in 100 ml of IMDM-5% FCS containing serial twofold dilution of the supernatant of G/S clones in 96-multiwell-plates. The maximum concentration of each supernatant is 50%. After 48 h the growth of NFS60 cells was determined by the MTT method and the results were compared with commercial rhG-CSF (Neupogen).
Results and discussion
High cell density cultivation of recombinant E. coli
Fed-batch culture of recombinant E. coli BL21 (DE3) harboring pET23a-g-csf vector was carried out in the glucose unlimited condition. Based on data obtained from previous experiments, an appropriate equation was correlated for decreasing of specific growth rate (Eq. 4) during feeding period, to avoid formation of growth inhibitory metabolites [15, 16].
where t is the time of fermentation and t 0 is the time of start feeding.
By substituting Eq. (4) into (3), glucose-feeding rate was determined. The final cell density was reached to ~138 g dry cell wt l−1 after 25 h cultivation by using this feeding strategy. Plasmid stability was approximately remained constant throughout the fermentation, and hG-CSF was slightly increased to 1.6 g hG-CSF l−1 in the end of the fermentation without induction (Fig. 1). Acetate concentration in the HCDC was below 3.1 g l−1 which is much lower than the reported growth-inhibitory concentration of acetate [23, 24]. In this research, HCDC were successfully obtained by just controlling the specific growth rate using exponential feeding. This approach is simple and efficient and does not need any special equipment or advance computer controller.

Fed-batch culture of recombinant E. coli BL21 (DE3) [pET23a-g-csf] using glucose as carbon and energy source in a 2-l bench top bioreactor containing 1 l of defined M9 modified medium. (Filled diamond) cell density, (filled triangle) specific growth rate and (filled square) plasmid stability. The arrow indicates start time of feeding
Production of hG-CSF in HCDC
In the fed-batch culture of recombinant E. coli BL21 (DE3) [pET3a-g-csf] at variable specific growth rate feeding strategy, the expression of hG-CSF was induced when cell density reached to ~48 g dry cell wt l−1, by addition of isopropyl-β-d-thiogalactopyranoside (IPTG) in the 3.0 mM final concentration. The maximum amount of hG-CSF was achieved after 4 h of post-induction (Fig. 2). The cell density and hG-CSF concentration were reached to about ~64 g dry cell wt l−1 and ~14 g hG-CSF l−1 at 19 h. The final specific yield and overall productivity of hG-CSF were obtained as 223 mg hG-CSF g−1 dry cell wt and 775 mg hG-CSF l−1 h−1, respectively.

The profile of recombinant hG-CSF and cell density in fed-batch culture of recombinant E. coli BL21 (DE3) [pET23a-g-csf] after induction with 3 mM IPTG using glucose as carbon and energy source in a 2-l bench top bioreactor containing 1 l of defined M9 modified medium. (Filled square) recombinant hG-CSF concentration, (filled triangle) cell density and (filled diamond) specific growth rate. The arrow indicates start time of feeding
Cell lysis and inclusion bodies preparation
The percentage of cell lysis is an important factor to produce native products. With this in mind, in order to do cell lysis we used high-pressure homogenizer at 800 and 1,200 bar pressure. Cell lysis was done according to procedure illustrated in Scheme 1. The efficiency of cell lysis was measured with plate culture of lysed cells suspension in LB agar and following colony counting. The highest efficiency is obtained after three rounds of homogenization at 1,200 bar (Table 1). Therefore, this condition was chosen for further research.
Washing of inclusion bodies
After cell lysis, two buffers containing Triton X-100 (1%) and 1% sarcosyl were chosen and used for removal of impurities from inclusion bodies. Washings were performed according to Scheme 1. After each step the suspension was centrifuged and the supernatant was analyzed with different analytical methods such as LAL test, ELISA, UV260 and Bradford method. Table 2 shows that after the first step about 58% of LPS was removed but by the third step we reached the plateau. These data show that Triton solubilizes the cell debris and accompanying LPS of inclusion bodies, which result in to appearance of lipopolysaccharides in the supernatant.
Further more, the amount of hG-CSF is decreased due to washing of inclusion bodies by about 2.3%, and the final yield of this step becomes 97.7%. However, this step has no effect on the removal of nucleic acid Impurities. At the next step, the insoluble inclusion bodies were washed with buffer Tris containing 1% of sarcosyl. The washing was performed following the procedure described in Scheme 1. After each wash the suspension was centrifuged and the supernatant was analyzed with different analytical methods. Table 2 does not show any change in the amount of nucleic acids and LPS for sarcosyl wash. In addition, the amount of hG-CSF during this process is decreased about 6% and final yield of this step is 94%.
Inclusion bodies solubilization
The wet pellet of IBs was dissolved according to procedure described in Scheme 1. After solubilization, the solution was centrifuged in 11,500 rpm for 30 min at 4 °C.
Refolding of rhG-CSF
The solubilized IB was refolded according to procedure as described in “Materials and methods” section. After dialysis, the solution containing 500 μg ml−1 protein was incubated over night at 4 °C to obtain the optimum amount of refolded protein. As seen in Table 3, the purity of hG-CSF in this step is 60%. The data show that about 35% of refolded protein was aggregated and LPS decreased to 50 EU. Clearance of endotoxin, nucleic acids and yield of process are shown in Table 3.
Cooper ion and other trace metal have been reported to catalyze air oxidation of proteins due to their ability to accelerate thiol oxidation at concentrations ranging from 0.1 to 10 μM [25]. Recombinant hG-CSF contains a free cystein at position 17 and two intramolecular disulfide bonds, Cys36–Cys42 and Cys64–Cys74. The two-disulfide bonds form two small loops, which are separated by 21 amino acids. Like other bacteria-derived recombinant proteins, recombinant hG-CSF produced in E. coli requires an oxidative folding procedure in order to recover its biological activity. In this paper to improve the condition for refolding of recombinant hG-CSF, we studied the effect of cupper concentration on aggregation of recombinant hG-CSF during refolding. For this purpose, different concentration of CuSO4 was added to refolding buffer and solubilized hG-CSF was dialyzed against refolding buffer. After refolding the aggregated form of protein was measured by A 340 nm [17]. The data show that the best CuSO4 concentration is 40 μM (Table 4). So, this condition was chosen for the further research.
DEAE-chromatography development
The DEAE column is involved in removal of impurities such as product-related impurities and host-cell impurities. Clearance of different host-cell impurities using DEAE column before refolding is shown in Table 3. Separations were performed according to procedure illustrated in Scheme 2. The solubilized IB was loaded on DEAE column and eluted with a linear gradient NaCl (0–1 M). The resulting fractions were analyzed by SDS-PAGE (see Fig. 3). Further DEAE pool samples were analyzed to measure the quantity of the product and step yield.

SDS-PAGE analysis of different purification step of rhG-CSF from E. coli. The commercial rhG-CSF (Neupogen), lane M washed inclusion body, lane 1 DEAE fraction, lane 2 Butyl fraction, lane 3. The gel was stained with Coomassie Brillient Blue R250 method and was and quantified by densitometry
Experiments were performed at pH 7.5, 8.0 and 8.5. At pH 8.0 the step yield increased to 80–90%, so, pH 8.0 was chosen as the optimum pH. Figure 3 shows the Coomassie brilliant R250 stained SDS-PAGE analysis of the relevant fractions.
Butyl-Sepharose chromatography-column development
The butyl column is primarily involved in removal of impurities. Clearance of endotoxin and nucleic acids are shown in Table 3. After DEAE column, separations were performed according to procedure illustrated in Scheme 3. The fractions containing G-CSF were pooled and ammonium sulfate to a final concentration of 0.25 M was added. Second, the solution was put on Butyl-Sepharose column equilibrated with 20 mM Tris–HCl, pH 8.0, and 0.25 M ammonium sulfate. The desired protein was eluted with a linear 0.25–0 M ammonium sulfate gradient in the same buffer. The purity of the purified rhG-CSF was assayed by SDS-PAGE. EDTA and PMSF were used as metal protease and serine protease inhibitors, respectively. The resulting fraction was analyzed by different analytical methods. The obtained results (Table 3) show that 86% of loaded G-CSF is eluted from this column and about 34% of endotoxin is absorbed.
The fractions containing purified hG-CSF from Butyl column were pooled and desalted by passage through a column of Sephadex G25 (2.2 cm × 90 cm), previously equilibrated in buffer 10 mM sodium acetate, pH 5.5. After desalting, sucrose was added to solution by final concentration 20 mg ml−1 and stored at 4 °C. The resulting fraction was analyzed by different analytical methods. The results show that 94% of loaded G-CSF is eluted from this column, endotoxin is less than 1 EU and purity is 99.9% and clinically acceptable.
The biological activity of purified protein was measured by NFS60 proliferation assay that gave 2 × 108 unit mg−1.
References
Riesenberg D, Guthke R (1999) High-cell-density cultivation of Escherichia coli. Curr Opin Biotechnol 2:380–384. doi:10.1016/S0958-1669(05)80142-9
Lee SY (1996) High cell-density culture of Escherichia coli. Trends Biotechnol 14:98–105. doi:10.1016/0167-7799(96)80930-9
Lim HK, Jung KH, Park DH, Chung SI (2000) Production characteristics of interferon-α using an L-arabinose promoter system in a high-cell-density culture. Appl Microbiol Biotechnol 53:201–208. doi:10.1007/s002530050009
Basu S, Dunn A, Ward A (2002) G-CSF: function and modes of action. Int J Mol Med 10:3–10
Hill CP, Osslund TD, Eisenberg D (1993) The structure of granulocyte-colony-stimulating factor and its relationship to other growth factors. Proc Natl Acad Sci USA 90:5167–5171. doi:10.1073/pnas.90.11.5167
Welte BK, Gabrilove J, Bronchud MH, Platzer E, Morstyn G (1996) Filgrastim (r-metHuG-CSF): the first 10 years. Blood 88(6):1907–1929
Kuga T, Komatsu Y, Yamasaki M, Sakine S, Miyaji H, Nishi T et al (1989) Mutagenesis of human granulocyte colony stimulating factor. Biochem Biophys Res Commun 159(1):103–111. doi:10.1016/0006-291X(89)92410-8
Wingfield P, Benedict R, Turcatti G, Allet B, Mermod JJ, DeLamarter J et al (1988) Characterization of recombinant-derived granulocyte-colony stimulating factor (G-CSF). Biochem J 256:213–218
Lu HS, Clogston CL, Narhi LO, Merewether LA, Pearl WR, Boone TC (1992) Folding and oxidation of recombinant human granulocyte colony stimulating factor produced in Escherichia coli. J Biol Chem 267(13):8770–8777
Komatsu Y, Matsumoto T, Kuga T, Nishi T, Sakine S, Saito A (1987) Cloning of granulocyte colony stimulating factor cDNA and its expression in Escherichia coli. Jpn J Cancer Res 78:1179–1181
Devlin PE, Drummond RJ, Toy P, Mark DF, Watt KW, Devlin JJ (1988) Alteration of amino-terminal codons of human granulocyte colony stimulating factor increases expression levels and allow efficient processing by methionine aminopeptidase in Escherichia coli. Gene 65:13–22. doi:10.1016/0378-1119(88)90412-X
Soo-Hyung K, Kyu-Heum N, Jang-Hyeon P, Choong-II P, Se-Yong L, Young Ik L (1995) High level expression and simple purification of recombinant human granulocyte colony stimulating factor in E. coli. Biotechnol Lett 17:687–692. doi:10.1007/BF00130351
Bishop B, Koay DC, Sartorelli AC, Regan L (2001) Reengineering granulocyte-colony stimulating factor (G-CSF) for enhanced stability. J Biol Chem 276:33465–33470. doi:10.1074/jbc.M104494200
Fallah MJ, Akbari B, Saeedinia AR, Karimi M, Zeinoddini M, Soleimani M (2003) Over expression of recombinant human granulocyte colony stimulating factor in E. coli. IJMS 28:131–134
Khalilzadeh R, Shojaosadati SA, Maghsoudi N, Mohammadian-Mosaabadi J, Mohammadi MR, Bahrami A et al (2004) Process development for production of recombinant human interferon-γ expressed in Escherichia coli. J Ind Microbiol Biotechnol 31:63–69. doi:10.1007/s10295-004-0117-x
Khalilzadeh R, Shojaosadati SA, Bahrami A, Maghsoudi N (2003) Over-expression of recombinant human interferon-gamma in high cell density fermentation of recombinant Escherichia coli. Biotechnol Lett 25:1989–1992. doi:10.1023/B:BILE.0000004390.98648.25
Mulkerrin MG, Wetzel R (1989) pH dependence of the reversible and irreversible thermal denaturation of gamma interferon. Biochemistry 28:6556–6561. doi:10.1021/bi00442a005
Seeger A, Schneppe B, McCarthy JEG, Deckwer W-D, Rinas U (1995) Comparison of temperature- and isopropyl-β-d-thiogalacto-pyranoside-induced synthesis of basic fibroblast growth factor in high-cell-density cultures of recombinant Escherichia coli. Enzyme Microb Technol 17:947–953. doi:10.1016/0141-0229(94)00123-9
Panda AK, Khan RH, Appa Rao KBC, Totey SM (1999) Kinetics of inclusion body production in batch and high cell density fed-batch culture of Escherichia coli expressing ovine growth hormone. J Biotechnol 75:161–172. doi:10.1016/S0168-1656(99)00157-1
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle protein dyebinding. Anal Biochem 72:248–254. doi:10.1016/0003-2697(76)90527-3
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi:10.1038/227680a0
Oshima Y, Tojo A, Niho Y, Asano S (2000) Biological activity of human granulocyte colony stimulating factor with a modified C-terminus. Biochem Biophys Res Commun 267:924–927. doi:10.1006/bbrc.1999.2062
Kleman GL, Strohl WR (1994) Development in high cell density and high productivity microbial fermentation. Curr Opin Biotechnol 5:180–186. doi:10.1016/S0958-1669(05)80033-3
Shiloach J, Kaufman J, Guillard AS, Fass R (1996) Effect of glucose supply strategy on acetate accumulation, growth, and recombinant protein production by Escherichia coli JM109. Biotechnol Bioeng 49:421–428. doi :10.1002/(SICI)1097-0290(19960220)49:4<421::AID-BIT9>3.0.CO;2-R
Saxena P, Wetlaufer DB (1970) Formation of three-dimensional structure in proteins. I. Rapid nonenzymatic activation of reduced lysozyme. Biochemistry 9:5015–5023. doi:10.1021/bi00827a028
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Khalilzadeh, R., Mohammadian-Mosaabadi, J., Bahrami, A. et al. Process development for production of human granulocyte-colony stimulating factor by high cell density cultivation of recombinant Escherichia coli . J Ind Microbiol Biotechnol 35, 1643–1650 (2008). https://doi.org/10.1007/s10295-008-0408-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-008-0408-8