
Abstract
Narcolepsy is a rare disease that entails excessive daytime sleepiness, often associated with sudden episodes of muscle weakness known as cataplexy. Narcolepsy with cataplexy (NC) is due to the loss of hypothalamic neurons that release the neuropeptides orexin A and B. Orexin neuron projections prominently target brain structures involved in wake-sleep state switching and the central autonomic network. This review provides an updated summary of the links between NC and autonomic cardiovascular dysfunction from a translational perspective. The available evidence suggests that, compared with control subjects, the heart rate in patients and animal models with NC is variable during wakefulness and normal to high during sleep. Responses of the heart rate to internal stimuli (arousal from sleep, leg movements during sleep, defense response) are blunted. These alterations result from orexin deficiency and, at least during wakefulness before sleep, involve decreased parasympathetic modulation of the heart rate. On the other hand, NC in patients and animal models is associated with a blunted fall in arterial blood pressure from wakefulness to sleep, and particularly to the REM state, coupled to a variable decrease in arterial blood pressure during wakefulness. The former effect is caused, at least in part, by deranged control of the heart, whereas the latter may be due to decreased vasoconstrictor sympathetic activity. Systematic studies are warranted to help clarify whether and how the links between NC and autonomic dysfunction impact on the cardiovascular risk of patients with narcolepsy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Narcolepsy entails excessive daytime sleepiness, often associated with sudden episodes of muscle weakness known as cataplexy. In 2000, human narcolepsy with cataplexy (NC) was linked to the loss of a restricted population of hypothalamic neurons that release the neuropeptides orexin A and B (OX-A/B, also called hypocretin 1 and 2). These neuropeptides had been discovered somewhat serendipitously by independent groups working on rats 2 years before [15, 54]. The discovery of the link between OX-A/B and human NC was heralded, in 1999, by demonstrations that mutations of orexin receptor 2 (OX-R2) cause familial NC in dogs [40], and that the phenotype of orexin knockout mice closely resembles that of NC patients [9].
This review aims to provide an updated summary of the links between narcolepsy and autonomic dysfunction. Although this topic has been the subject of several recent reviews [7, 27, 39, 52, 64], the present paper is different in that it focuses on cardiovascular autonomic control from a translational perspective.
The first section of the review will summarize key background information on narcolepsy and the orexin system. The second and third sections will provide an updated overview of the reported links between narcolepsy and autonomic dysfunction, with a focus on derangements of the heart rate (HR) and arterial blood pressure (ABP) during sleep in patients and animal models with NC. The last section will discuss the implications of the previous sections for research into the links between narcolepsy and autonomic dysfunction.
What is narcolepsy?
Narcolepsy is currently diagnosed based on excessive daytime sleepiness, with daily periods of an irrepressible need to sleep, short sleep latency, and periods of rapid-eye-movement (REM) sleep at sleep onset [44]. Narcolepsy is further classified into two types, identified as narcolepsy type 1 and 2 (NT1/2). A diagnosis of NT1 requires the occurrence of either cataplexy or an OX-A concentration in the cerebrospinal fluid (CSF) <1/3 that seen in healthy subjects. NT2 is diagnosed in those who meet the criteria for hypersomnia but do not have cataplexy or a low OX-A concentration in the CSF. In this review, the term NC will be applied to refer to narcolepsy with cataplexy in animal models and human subjects, the latter irrespective of whether diagnosis preceded or followed the publication of the NT1/NT2 criteria [44]. The general term “narcolepsy” will be applied to refer to narcolepsy irrespective of the occurrence of cataplexy and/or orexin deficiency.
The exact prevalence of NT1 and NT2 is still unclear. The overall prevalence of narcolepsy is estimated at between 25 and 50 per 100,000 [42], which meets the definition of a rare disease [47]. The incidence of narcolepsy has a major peak between 15 and 30 years of age [42, 75]. Narcolepsy also occurs in children, and its incidence in those aged 5–19 years has risen significantly in northern Europe since the recent influenza pandemic [75]. Growing evidence points to specific clinical features of NC in childhood, including rapid weight gain and precocious puberty [53]. On the other hand, narcolepsy is a life-long disease [37], and it is still unclear whether it entails specific clinical features in older age.
NC is accompanied by a wide and heterogeneous spectrum of comorbidities. NC entails a moderate tendency to obesity [30], which, as previously mentioned, is may be particularly prominent during childhood [53]. Although the available evidence is still not conclusive (see for example [73] in relation to restless legs syndrome), it suggests that NC also entails an increased rate of occurrence of obstructive sleep apneas [56] and periodic leg movements during wakefulness and sleep [13] as well as an increased prevalence of restless legs syndrome [51]. There is also evidence that NC is associated with arterial hypertension [45], particularly in elderly patients [37], and with a lower than normal nightly decrease in ABP, i.e., a nondipper ABP profile [12, 25, 62]. Interestingly, recent evidence suggests that the perioperative outcomes of patients with narcolepsy undergoing surgery are more frequently complicated by emergency response team activations due to hemodynamic instability than those of matched control subjects [8].
In the general population, obesity is a powerful cardiovascular risk factor (particularly if it starts during childhood [1]), as are obstructive sleep apneas [19] and a nondipper ABP profile [32]. Evidence is actually accruing that the nocturnal values of ABP play the key role in the pathophysiology of ABP-related morbidity and mortality [33]. There is also evidence that periodic leg movements during sleep and restless legs syndrome increase cardiovascular risk in non-narcoleptic patients [77]. Tobacco smoking is more frequent in patients with NC than in control subjects [2]. The extent to which the combination of these factors increases cardiovascular risk and mortality in patients with narcolepsy with or without cataplexy is still a matter of debate [34, 46].
Pharmacological therapy for narcolepsy is symptomatic and based on several treatment options, including sodium oxybate to stabilize nocturnal sleep, reduce daytime sleepiness, and limit cataplexy; stimulants such as modafinil, methylphenidate, and amphetamine to prevent excessive daytime sleepiness; and low-dose antidepressants such as venlafaxine, a serotonergic and noradrenergic reuptake inhibitor, to prevent cataplexy. In addition, antagonists of the histamine H3 receptor are a recently developed therapeutic option to promote wakefulness in patients with narcolepsy [5].
The orexin system and narcolepsy
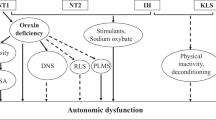
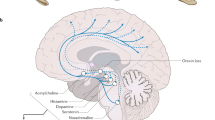
As previously mentioned, a finding of low CSF values of OX-A is one of the diagnostic criteria for NT1 [44]. Orexins are released selectively by a population of neurons localized in a restricted area of the tuberal region of the hypothalamus, caudal to the paraventricular nucleus [50]. Orexin neurons project widely to the entire central nervous system, prominently targeting brain structures involved in wake-sleep state switching [49, 57] and the central autonomic network [67] (Fig. 1). Through these projections, orexin neurons may thus play a leading role in modulating wake-sleep states and their effects on autonomic control [69] (Fig. 2).

Reproduced from [27] with permission
Schematic diagram showing a sagittal section of the human brainstem and diencephalon with the main projections from orexin (OX) neurons to components of the central autonomic network. A5 homonymous noradrenergic cell group of the pons, DMV dorsal motor nucleus of the vagus nerve, mRN medullary raphe nuclei, NAmb nucleus ambiguus, NTS nucleus of the solitary tract, PVN paraventricular nucleus of the hypothalamus, RVLM rostral ventrolateral medulla, RVMM rostral ventromedial medulla

Modified from [69] with permission
Schematic diagram showing the general central mechanisms by which orexin neurons may modulate both wake-sleep states and their effects on cardiovascular control. Please refer to the text for further details. NTS nucleus of the solitary tract
Orexin neurons co-release dynorphin and glutamate, and potentially also nociceptin/orphanin FQ, galanin, neurotensin, and γ-aminobutyric acid (GABA) [6]. All of these cotransmitters are likely to be lost in NC, together with orexins [11, 49]. The pathophysiological basis of NT2 is much less clear. Experiments on rats indicate that a loss of approximately 75% of orexin neurons causes only a 50% decrease in orexin CSF levels [22], which would not meet the criteria for diagnosing NT1 in humans [44]. It is therefore possible that a partial loss of orexin neurons underlies at least some of the cases of NT2, i.e., narcolepsy without cataplexy and with preserved CSF orexin levels.
OX-A and OX-B are produced by proteolytic cleavage of a precursor protein, and they bind two excitatory metabotropic receptors named orexin receptor 1 (OX-R1) and OX-R2 [23]. The ligands OX-A and OX-B are usually considered to have equal affinities for OX-R2, whereas OX-B is thought to be a much weaker ligand than OX-A for OX-R1 [38].
The full clinical features of narcolepsy, or at least NC, likely result from the loss of orexin binding to both orexin receptors, as well as from the loss of the multiple transmitters co-released by the orexin neurons. Nonetheless, as previously mentioned, orexin knockout mice, whose mutation prevents the production of OX-A/B but not that of the orexin cotransmitters, strikingly recapitulate the clinical phenotype of patients with NC [9]. Moreover, genetic mutations leading to the loss of OX-R2 are sufficient to cause core signs of NC in dogs [40] and mice [76]. Taken together, these pieces of evidence from animal models indicate that the key driving event in the pathophysiology of NC is the loss of OX-A/B binding to OX-R2.
Orexin neurons are wake-active, exhibiting a slow tonic firing during wakefulness that ceases during non-REM and REM sleep, with the exception of transient discharges during the latter state [72]. The estimated extracellular concentration of OX-A is in the nanomolar range [78], approximately one order of magnitude lower than the concentration required for the half-maximal response (EC50) of orexin receptors [54]. During wakefulness, the orexin neuron discharge is enhanced by appetitive stimuli [29]. On the other hand, brain extracellular levels of OX-A depend loosely, if at all, on the wake-sleep state in rats [78] and cats [36]. OX-A levels in the lumbar CSF are paradoxically higher at the end of the night than in the afternoon in healthy subjects [24]. The mechanisms underlying this apparent dissociation between the state dependency of orexin neuron firing and extracellular OX-A levels have not been elucidated. However, this dissociation may explain specific clinical features of NC that occur during sleep, such as obstructive sleep apneas [19], periodic leg movements during sleep [77], and a nondipper ABP profile [12, 25, 62].
The orexins and their receptors have been detected based on mRNA and/or immunohistochemistry outside the hypothalamus in a wide range of tissues, including the intestine, pancreas, adrenals, kidney, adipose tissue, and reproductive tract [31]. Recent data suggest that the OX-R2 is expressed by cardiomyocytes and regulates cardiac function, potentially representing a therapeutic target for the treatment of heart failure [48]. Nonetheless, the sources of orexins outside the central nervous system and their physiological significance are still a matter of controversy [38], and their involvement in the pathophysiology of narcolepsy is still unexplored.
Narcolepsy, the orexin system, and heart rate control
Heart rate control in narcoleptic patients with cataplexy
Detailed measurements of HR in patients with NC vs. control subjects have yielded inconsistent results, spanning from no significant differences between patients and controls [12, 17, 21] to decreased HR in patients during daytime wakefulness [18] and increased HR in patients, particularly during sleep [25, 71, 74] (Table 1).
Somewhat more consistent results have been produced by studies of heart rate variability (HRV) in patients with NC (Table 1). Two studies reported alterations of HRV in patients with narcolepsy compared with control subjects selectively during wakefulness before sleep: an increased ratio of low-frequency to high-frequency power of HRV (LF/HF) in patients with narcolepsy [20], and a decreased root mean square of successive interbeat intervals (RMSSD) in patients with NC [70]. Another study found increased values of LF/HF HRV in NC patients during morning wakefulness, with no further increase during the head-up tilt test [26]. Conversely, HRV was found to be higher in patients with NC than in control subjects during wakefulness in the morning [21]. During sleep, the HRV associated with leg movements and arousals [14, 71] was found to be reduced in patients with NC. However, indirect estimates of the central autonomic control of the heart based on the relationship between spontaneous fluctuations of the interbeat interval and ABP were found to be either preserved or increased during sleep in patients with NC compared with control subjects, potentially because of the increased occurrence rate of leg movements during sleep in these patients [70].
All the clinical studies mentioned in this section were performed on adult subjects, and, with the exception of two studies [20, 71], they explicitly focused on patients with NC. The occurrence of alterations in the mean value of HR or HRV in children with NC and in patients of any age with NT2 (i.e., narcolepsy without cataplexy and/or with a normal CSF orexin concentration) thus warrants further studies. For the latter, the limited available evidence suggests that orexin deficiency is associated with more severe alterations of HRV in adult patients with narcolepsy [71].
Heart rate control in animal models of narcolepsy with cataplexy
Studies of animal models of narcolepsy have focused on models of NC with orexin deficiency and have mainly been performed on mice. These studies have also yielded conflicting results on alterations in the control of HR compared with control animals. One potential confounder in these studies is genetic differences (the “genetic background”) other than the specific mutation(s) that cause orexin deficiency. To control the confounding effects of genetic background, most studies are performed on inbred mouse strains that have been mated using a suitable breeding scheme (usually brother × sister) for ≥20 consecutive generations. As a result, the genetic backgrounds of all the mice of a given inbred strain are essentially identical (“pure”), and differ from those of mice of other inbred strains. Inbred strains are identified by unique combinations of letters and numbers, such as C57BL/6, 129/SvEv, and DBA/2. However, the technical procedures for generating novel mouse models often yield hybrid mice with variable contributions of genes from two different mouse strains (i.e., a “mixed” genetic background). Thus, it is not uncommon for early studies on a new mouse mutant to be performed on hybrid mice, even though this affords less efficient control of genetic confounders.
An early report on orexin knockout mice on a mixed C57BL/6–129/SvEv genetic background found no significant difference in HR compared with wild-type mice throughout the light–dark cycle, but a weaker HR increase during the defense response, either artificial (stimulation/disinhibition of the perifornical area) or spontaneous (resident-intruder test) [35]. These results were essentially replicated by the same group on transgenic orexin-ataxin-3 mice on a mixed C57BL/6–DBA/2 genetic background, in which expression of the neurotoxin ataxin-3 kills orexin neurons after birth [79]. No significant difference in HR during wakefulness and sleep was also reported by another group on orexin-ataxin-3 rats [58].
These results were partly challenged by a series of experiments performed by our group. We found that orexin knockout mice on a pure C57BL/6 genetic background had higher HR than wild-type mice during wakefulness, non-REM sleep, and REM sleep [3]. In the same study, we found that orexin-ataxin-3 mice on a pure C57BL/6 genetic background had higher HR than wild-type mice during REM sleep, whereas this difference was not significant for orexin ataxin-3 mice on a mixed C57BL/6–DBA/2 genetic background [3]. We performed these experiments on young-adult (approx. 15 weeks of age) mice at an ambient temperature of 25 °C. These experimental details may be relevant in light of later findings from our laboratory that young-adult orexin-ataxin-3 mice on a mixed C57BL/6–DBA/2 genetic background have values of HR similar to those of wild-type mice at an ambient temperature of 20 °C, but values higher than those of wild-type mice at an ambient temperature of 30 °C [41]. We also found that middle-aged (10–11 months of age) orexin-ataxin-3 mice on a pure C57BL/6 genetic background had higher values of HR during each wake-sleep state and reduced HRV during non-REM sleep compared with wild-type mice [65].
In mice with NC, we also found subtle alterations in HRV, which mostly consisted of weaker baroreflex changes in interbeat interval during REM sleep, associated with physiological events such as spontaneous increases in ABP [41, 65, 66]. In addition, in correspondence with spontaneous surges of ABP during non-REM sleep, young-adult orexin-ataxin-3 mice on a mixed genetic background showed weaker baroreflex changes in interbeat interval at an ambient temperature of 30 °C [41], whereas middle-aged orexin-ataxin-3 mice on a pure C57BL/6 genetic background showed weaker central autonomic control of the interbeat interval [65].
Overview of the link between narcolepsy with cataplexy and heart rate control: a translational perspective
As highlighted in the previous sections, the association between NC and alterations of HR control is a matter of controversy and inconsistency. The results of the published reports on this matter may be categorized into three groups. The first group consists of only one study in NC patients during wakefulness, which reported lower values of HR than in control subjects [18]. The second group consists of studies on patients [12, 17, 20, 21] and animal models [3, 35, 58, 79] with NC, which reported values of HR similar to those of control subjects. The last group consisted of studies on patients [25, 71, 74] or animal models [3, 41, 65] with NC, which reported values of HR higher than those in control subjects, particularly during sleep. This last phenomenon was also observed for orexin knockout mice [3], which lack orexin peptides with preserved hypothalamic neurons [9], meaning that it is caused by a lack of orexin peptides rather than a lack of orexin cotransmitters.
Although not yet tested systematically, genetic background [3, 41], ambient temperature [41], and age [65] may be significant modulators of the HR phenotype associated with an orexin deficiency in mice, with milder HR derangements seen in younger mice, in those with a mixed genetic background, and at colder ambient temperatures. Weaker HR responses to internal stimuli are a frequent finding in mouse models (defense reaction [35, 79]) and patients (responses to arousals and leg movements [14, 71]) with NC. Subtle alterations of HRV were reported in mouse models [41, 65, 66] and patients [20, 21, 70] with NC, but the findings for animal models did not mirror those for patients.
Potential mechanisms of heart rate control dysfunction in narcolepsy with cataplexy
The mechanisms responsible for the reported alterations in the mean values of HR and HRV in patients with NC are still unclear. Increases in the mean value of HR in patients and mice with NC [3, 25, 41, 65, 71, 74] may be caused by increased sympathetic tone to the heart, by decreased parasympathetic tone to the heart, and, possibly, by increased intrinsic HR [67]. Recent experimental data [43] question the interpretation of the LF/HF index, which was found to be increased during wakefulness in patients with narcolepsy [20, 26]. Nonetheless, the decrease in the RMSSD index reported during wakefulness before sleep in patients with NC [70] is strongly suggestive of a decrease in cardiac parasympathetic modulation [67]. Accordingly, spontaneous cardiac baroreflex sensitivity (cBRS), which is highly dependent on cardiac parasympathetic modulation [68], is also decreased during wakefulness before sleep in patients with NC compared with control subjects [70]. Spontaneous cBRS is otherwise preserved in patients with NC during sleep [70] and in animal models of NC during each wake-sleep state [41, 65, 66, 79], although one study on transgenic orexin-ataxin-3 rats reported reduced cBRS during REM sleep [58]. The available data, therefore, do not generally support the association of significant deficits in baroreflex control or cardiac vagal modulation with NC. Further studies are needed to test whether this association is specifically present during wakefulness before sleep.
We have previously hypothesized that the blunting of cardiac parasympathetic modulation selectively during wakefulness before sleep in patients with NC [70] may be due to cognitive components associated with the fight against somnolence [80]. On the other hand, the increases in HR during the artificial defense response, which were found to be blunted in orexin-ataxin-3 mice compared with wild-type control mice, were abolished by beta-adrenergic blockade [79]. A blunted sympathetic modulation of HR in NC may thus underlie the blunted HR component of the defense reaction [35, 79], and may help to explain the blunted HR responses to internal stimuli such as arousals and leg movements [14, 71] and spontaneous increases in ABP [41, 65, 66].
Basic research on the effects of orexins on HR has provided insight into the mechanisms of the reported alterations of HR in human subjects and animal models with NC. Early reports indicated that orexin injection in the lateral cerebral ventricle increases HR in unanesthetized rats, with greater effects seen for OX-A than for OX-B [61]. These effects of orexins may underlie the reduced HR occasionally observed in patients with NC during wakefulness [18], but not the findings of increased HR in NC patients and animal models [3, 25, 41, 65, 74].
Nonetheless, the reported increases in HR after orexin injection in the cerebral ventricles likely reflect effects of orexins on multiple brain structures (Fig. 1) and at concentrations several orders of magnitude higher than physiological concentrations [61, 78]. These issues may be relevant because localized microinjection of orexins in the nucleus of the solitary tract, which is a key structure of the central autonomic network controlling HR [67], decreases HR at lower concentrations and increases HR at higher concentrations [60]. OX-A microinjection in the nucleus ambiguus, which is the main site of the preganglionic parasympathetic neurons that control HR [67], also decreases HR by increasing cardiac parasympathetic activity [10]. This suggests that OX-A exerts a cardiac vagotonic effect, at least at the level of the nucleus ambiguus. Thus, orexin deficiency in patients with NC would imply a lack of this vagotonic effect, potentially explaining why patients with NC may show increased values of HR [3, 25, 41, 65, 74] and reduced cardiac vagal modulation [70] compared with control subjects.
Narcolepsy with cataplexy, the orexin system, and the control of arterial blood pressure
Control of arterial blood pressure in narcoleptic patients with cataplexy
The control of ABP in patients with narcolepsy during wakefulness and sleep has long remained a relatively neglected topic, with only one published study (from 1986), which reported normal values of ABP during wakefulness and sleep in patients with NC [28]. This conclusion was first challenged in 2012 by a careful study performed under controlled conditions with noninvasive beat-to-beat measurement of ABP. This study reported a nondipper ABP profile (i.e., a lower than normal decrease in ABP from daytime to nighttime) in patients with NC compared with normal control subjects. However, the absolute values of ABP were significantly higher in patients with NC than in control subjects only during REM sleep [25]. Another study published the same year on a larger sample of patients with NC in which ambulatory ABP monitoring was employed reported a nondipper profile of diastolic ABP in 31% of the patients vs. 3% of healthy controls, but no significant difference between groups in the absolute values of diastolic ABP during daytime and nighttime [12]. These discrepancies suggest that patients with NC differ from control subjects in the ability to modulate ABP, but these differences may be obscured by relatively wide between-subject differences in the 24-h average values of ABP.
Further support for this conclusion was provided by a later study of patients with NC employing beat-to-beat ABP monitoring, which reported that the relative decrease in ABP from wakefulness to non-REM sleep was significantly less pronounced in NC patients than in healthy controls, whereas the absolute values of ABP did not differ significantly between groups [17]. The frequent occurrence of a nondipper ABP pattern in patients with NC was further confirmed recently by a study that estimated ABP based on pulse transit time [62]. At variance with these results, one study reported lower values of ABP in patients with NC during wakefulness compared with control subjects [18].
On the other hand, the investigation of beat-to-beat variability of ABP within each wake-sleep state has been relatively neglected in patients with narcolepsy, with one study [21] reporting increased ABP variability during morning wakefulness, and another study [70] reporting no significant differences during wakefulness and sleep in NC patients compared with healthy controls. The clinical studies mentioned in this section are summarized in Table 2. None of these studies focused on children with NC or on patients of any age with NT2.
Control of arterial blood pressure in animal models of narcolepsy with cataplexy
Studies on the control of ABP in animal models of NC yielded results that were broadly consistent with those obtained for patients. An early study on orexin knockout mice on a mixed C57BL/6–129/SvEv genetic background reported significantly lower values of ABP than in wild-type control mice during the light period and the dark period, and a weaker ABP increase during the defense response [35]. These results were essentially confirmed by the same group for transgenic orexin-ataxin-3 mice on a mixed C57BL/6–DBA/2 genetic background [79]. Similar results were later obtained by an independent group for orexin-ataxin-3 rats, which showed lower values of ABP than wild-type controls during wakefulness and sleep, while the relative changes in ABP across wake-sleep transitions were preserved [58].
As also seen for the results on HR control, these conclusions regarding the control of ABP in mouse models of NC were partly challenged by a series of experiments performed by our group. In particular, we found that young-adult orexin knockout mice and orexin-ataxin-3 mice on a pure C57BL/6 genetic background had higher absolute values of ABP than wild-type controls during non-REM sleep and REM sleep, whereas young-adult orexin-ataxin-3 mice on a mixed C57BL/6–DBA/2 genetic background had higher absolute values of ABP than wild-type controls during REM sleep only. In each of these three strains, however, the relative decreases in ABP during either non-REM sleep or REM sleep compared to wakefulness were significantly less pronounced than those in wild-type control mice [3].
We further confirmed the occurrence of blunted relative decreases in ABP during sleep at ambient temperatures of 20 and 30 °C in young-adult orexin-ataxin-3 mice with a mixed C57BL/6–DBA/2 genetic background [41]. In that study, however, the absolute values of ABP during wakefulness were actually lower in orexin-ataxin-3 mice than in wild-type controls during wakefulness at an ambient temperature of either 20 or 30 °C, whereas the absolute values of ABP during sleep did not differ between strains [41]. The reasons for the discrepancy in the absolute values of ABP of NC mice between that study [41] and our previous report on young-adult orexin-ataxin-3 mice on the same mixed C57BL/6–DBA/2 genetic background [3] may lie in subtle differences in the experimental protocol, including a significantly longer postoperative recovery and the exposure to changes in ambient temperatures [41]. On the other hand, it is worth remarking that both studies [3, 41] consistently supported the occurrence of blunted relative changes in ABP between wakefulness and the sleep states in mouse models of NC. A later study by our group also reported blunted sleep-related differences of ABP in middle-aged orexin-ataxin-3 mice with a pure C57BL/6 genetic background at an ambient temperature of 25 °C compared with wild-type controls, while the absolute values of ABP did not differ significantly [65].
Finally, the beat-to-beat variability of ABP within each wake-sleep state was investigated in three studies from our group [41, 65, 66]. These studies did not detect any significant difference in the beat-to-beat variability of ABP within each wake-sleep state between either orexin knockout or orexin-ataxin-3 mice and wild-type controls.
Overview of the link between narcolepsy and the control of arterial blood pressure: a translational perspective
Taken together, these results on patients and animal models suggest that NC is associated with a blunted fall of ABP from wakefulness to sleep, and particularly to the REM state, coupled to a variable decrease in ABP during wakefulness, likely depending on genetic background and other unidentified environmental or stress-related factors. As a result, NC may be associated with ABP values that are lower than normal in wakefulness [18] and normal during sleep [41], or normal in wakefulness and higher than normal during sleep [17], particularly during REM sleep [3, 25, 65] (Fig. 3). Since sleep mainly occurs during the nighttime in human subjects, blunted differences in ABP between wakefulness and sleep may explain, at least in part, the frequent occurrence of a nondipper ABP profile in patients with NC [12, 25, 62]. On the other hand, the beat-to-beat variability of ABP within each wake-sleep state does not appear to vary between human subjects [70] or animal models [41, 65, 66] with NC and control subjects.

Schematic diagram showing the alterations of the control of arterial blood pressure (ABP) that may be associated with orexin deficiency in animal models and patients with narcolepsy. In control subjects (left), ABP physiologically decreases from wakefulness (W) to non-rapid-eye-movement sleep (NREM), and rises again during rapid-eye-movement sleep (REM). Orexin deficiency (right) entails a variable decrease in ABP during W (single arrow) coupled to a blunted decrease in ABP from W to NREM and (particularly) to REM (double arrows). As a result, orexin deficiency may be associated with ABP values that are lower than normal in W and normal during sleep (a) or normal in W and higher than normal during sleep (b)
Potential mechanisms of arterial blood pressure control dysfunction in narcolepsy with cataplexy
As is the case for alterations of HR, the mechanisms behind the reported alterations of ABP in patients and animal models with NC are still uncertain. The occurrence of lower values of ABP in either orexin knockout [35] or orexin-ataxin-3 [79] mice was abolished by prazosin, which blocks alpha-1 adrenergic receptors. This suggests that lower values of ABP in mice with NC are caused by lower sympathetic activity to blood vessels. Accordingly, the study of patients with NC that reported lower values of ABP during wakefulness also reported lower values of muscle sympathetic nerve activity in these patients [18].
In a different study, the blunted relative decrease in ABP from wakefulness to non-REM sleep in patients with NC was associated with a blunted relative decrease in HR, whereas the relative decreases in muscle and skin sympathetic nerve activities during sleep were preserved in these patients [17]. This mismatch between sleep-related changes in HR and sympathetic nerve activity to peripheral vascular beds cannot be parsimoniously explained based on baroreflex resetting, which, moreover, has never been directly demonstrated in patients or animal models with NC. Nonetheless, in control subjects, the physiological decrease in HR from wakefulness to non-REM sleep may help to decrease ABP by decreasing cardiac output [63]. Thus, the association between NC and a blunted decrease in ABP from wakefulness to non-REM sleep may be driven, at least in part, by a blunted sleep-related decrease in HR, implicating abnormally high values of HR during sleep.
Both decreases in ABP during wakefulness [35] and blunted differences in ABP between wakefulness and sleep [3] have been reported in orexin knockout mice with preserved hypothalamic neurons. The occurrence of relatively high values of ABP during sleep, and particularly in REM sleep, compared to wakefulness is preserved in double-mutant mice lacking orexin neurons and histamine transmission [4]. This suggests that the key mechanism at stake is the loss of orexin transmission, as opposed to the loss of histamine or of transmitters co-released by the orexin neurons.
Similarly to what was previously discussed for HR, injections of OX-B and (particularly) OX-A in the lateral cerebral ventricle increase ABP in anesthetized rats [55, 61]. Moreover, OX-A microinjection in the rostral ventrolateral medulla, a key site of presympathetic neurons controlling the heart and blood vessels [67] (Fig. 1), increases ABP by means of sympathetic activation elicited by both OX-R1 and OX-R2 binding [59]. A lack of these sympathoexcitatory effects of orexins may explain the occurrence of decreased ABP in patients [18] and animal models [35, 41, 58, 79] of NC, particularly during wakefulness.
On the other hand, orexins may exert bidirectional effects on ABP acting on the nucleus of the solitary tract [60] and the paraventricular nucleus of the hypothalamus [16] (Fig. 1), depending on concentration [60] and, potentially, on GABAergic cotransmission [16]. The available evidence suggests that the nucleus of the solitary tract and the paraventricular nucleus of the hypothalamus play leading roles in driving the effects of sleep states on ABP [69]. The association between orexin deficiency and blunted sleep-related differences in ABP may thus be due to the loss of the physiological modulation exerted by orexin receptors on the central circuits that shape autonomic outflow during sleep [69].
Conclusions and suggestions for a research agenda
The theme of research on the links between narcolepsy, the orexin system, and autonomic dysfunction is a substantial variability between the reported results. On the one hand, this highlights the need for systematic reviews and meta-analyses, which are still lacking in this field. On the other hand, this variability is difficult to explain given the relative rarity of narcolepsy and the technical difficulties associated with autonomic investigation during spontaneous behavior. Rather, this variability suggests the existence of one or more biological modifiers of the effects of orexins on cardiovascular control, which may include genetic, environmental (e.g., ambient temperature, stress), and developmental (i.e., age-related) factors [64].
There are limited data on cardiovascular control in elderly patients with NC and in patients of any age with narcolepsy without cataplexy, and virtually no data on narcoleptic children. Potential differences in cardiovascular control between NC and narcolepsy without cataplexy also deserve closer scrutiny, as do the effects of narcolepsy medications on cardiovascular control. Taking into account the impact of obesity, sleep-disordered breathing, periodic leg movements during sleep, and tobacco smoking may help to clarify whether and how the links between narcolepsy and autonomic cardiovascular dysfunction impact on the cardiovascular risk of patients with narcolepsy.
References
Ayer J, Charakida M, Deanfield JE, Celermajer DS (2015) Lifetime risk: childhood obesity and cardiovascular risk. Eur Heart J 36:1371–1376
Barateau L, Jaussent I, Lopez R, Boutrel B, Leu-Semenescu S, Arnulf I, Dauvilliers Y (2016) Smoking, alcohol, drug use, abuse and dependence in narcolepsy and idiopathic hypersomnia: a case-control study. Sleep 39:573–580
Bastianini S, Silvani A, Berteotti C, Elghozi JL, Franzini C, Lenzi P, Lo Martire V, Zoccoli G (2011) Sleep related changes in blood pressure in hypocretin-deficient narcoleptic mice. Sleep 34:213–218
Bastianini S, Silvani A, Berteotti C, Lo Martire V, Cohen G, Ohtsu H, Lin JS, Zoccoli G (2015) Histamine transmission modulates the phenotype of murine narcolepsy caused by orexin neuron deficiency. PLoS One 10:e0140520
Black SW, Yamanaka A, Kilduff TS (2017) Challenges in the development of therapeutics for narcolepsy. Progr Neurobiol 152:89–113
Bonnavion P, Mickelsen LE, Fujita A, de Lecea L, Jackson AC (2016) Hubs and spokes of the lateral hypothalamus: cell types, circuits and behaviour. J Physiol 594:6443–6462
Calandra-Buonaura G, Provini F, Guaraldi P, Plazzi G, Cortelli P (2016) Cardiovascular autonomic dysfunctions and sleep disorders. Sleep Med Rev 26:43–56
Cavalcante AN, Hofer RE, Tippmann-Peikert M, Sprung J, Weingarten TN (2017) Perioperative risks of narcolepsy in patients undergoing general anesthesia: a case-control study. J Clin Anesth 41:120–125
Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M (1999) Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98:437–451
Ciriello J, de Oliveira CV (2003) Cardiac effects of hypocretin-1 in nucleus ambiguus. Am J Physiol Regul Integr Comp Physiol 284:R1611–R1620
Crocker A, Espana RA, Papadopoulou M, Saper CB, Faraco J, Sakurai T, Honda M, Mignot E, Scammell TE (2005) Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology 65:1184–1188
Dauvilliers Y, Jaussent I, Krams B, Scholz S, Lado S, Levy P, Pepin JL (2012) Non-dipping blood pressure profile in narcolepsy with cataplexy. PLoS One 7:e38977
Dauvilliers Y, Pennestri MH, Petit D, Dang-Vu T, Lavigne G, Montplaisir J (2007) Periodic leg movements during sleep and wakefulness in narcolepsy. J Sleep Res 16:333–339
Dauvilliers Y, Pennestri MH, Whittom S, Lanfranchi PA, Montplaisir JY (2011) Autonomic response to periodic leg movements during sleep in narcolepsy-cataplexy. Sleep 34:219–223
de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS II, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG (1998) The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Nat Acad Sci USA 95:322–327
Dergacheva O, Yamanaka A, Schwartz AR, Polotsky VY, Mendelowitz D (2017) Optogenetic identification of hypothalamic orexin neuron projections to paraventricular spinally projecting neurons. Am J Physiol Heart Circ Physiol 312:H808–H817
Donadio V, Liguori R, Vandi S, Giannoccaro MP, Pizza F, Leta V, Plazzi G (2014) Sympathetic and cardiovascular changes during sleep in narcolepsy with cataplexy patients. Sleep Med 15:315–321
Donadio V, Liguori R, Vandi S, Pizza F, Dauvilliers Y, Leta V, Giannoccaro MP, Baruzzi A, Plazzi G (2014) Lower wake resting sympathetic and cardiovascular activities in narcolepsy with cataplexy. Neurology 83:1080–1086
Dong JY, Zhang YH, Qin LQ (2013) Obstructive sleep apnea and cardiovascular risk: meta-analysis of prospective cohort studies. Atherosclerosis 229:489–495
Ferini-Strambi L, Spera A, Oldani A, Zucconi M, Bianchi A, Cerutti S, Smirne S (1997) Autonomic function in narcolepsy: power spectrum analysis of heart rate variability. J Neurol 244:252–255
Fronczek R, Overeem S, Reijntjes R, Lammers GJ, van Dijk JG, Pijl H (2008) Increased heart rate variability but normal resting metabolic rate in hypocretin/orexin-deficient human narcolepsy. J Clin Sleep Med 4:248–254
Gerashchenko D, Murillo-Rodriguez E, Lin L, Xu M, Hallett L, Nishino S, Mignot E, Shiromani PJ (2003) Relationship between CSF hypocretin levels and hypocretin neuronal loss. Exp Neurol 184:1010–1016
Gotter AL, Webber AL, Coleman PJ, Renger JJ, Winrow CJ (2012) International Union of Basic and Clinical Pharmacology. LXXXVI. Orexin receptor function, nomenclature and pharmacology. Pharmacol Rev 64:389–420
Grady SP, Nishino S, Czeisler CA, Hepner D, Scammell TE (2006) Diurnal variation in CSF orexin-A in healthy male subjects. Sleep 29:295–297
Grimaldi D, Calandra-Buonaura G, Provini F, Agati P, Pierangeli G, Franceschini C, Barletta G, Plazzi G, Montagna P, Cortelli P (2012) Abnormal sleep-cardiovascular system interaction in narcolepsy with cataplexy: effects of hypocretin deficiency in humans. Sleep 35:519–528
Grimaldi D, Pierangeli G, Barletta G, Terlizzi R, Plazzi G, Cevoli S, Franceschini C, Montagna P, Cortelli P (2010) Spectral analysis of heart rate variability reveals an enhanced sympathetic activity in narcolepsy with cataplexy. Clin Neurophysiol 121:1142–1147
Grimaldi D, Silvani A, Benarroch EE, Cortelli P (2014) Orexin/hypocretin system and autonomic control: new insights and clinical correlations. Neurology 82:271–278
Guilleminault C, Salva MA, Mancuso J, Hayes B (1986) Narcolepsy, cataplexy, heart rate, and blood pressure. Sleep 9:222–226
Hassani OK, Krause MR, Mainville L, Cordova CA, Jones BE (2016) Orexin neurons respond differentially to auditory cues associated with appetitive versus aversive outcomes. J Neurosci 36:1747–1757
Heier MS, Jansson TS, Gautvik KM (2011) Cerebrospinal fluid hypocretin 1 deficiency, overweight, and metabolic dysregulation in patients with narcolepsy. J Clin Sleep Med 7:653–658
Heinonen MV, Purhonen AK, Makela KA, Herzig KH (2008) Functions of orexins in peripheral tissues. Acta Physiol 192:471–485
Hermida RC, Ayala DE, Mojon A, Fernandez JR (2013) Blunted sleep-time relative blood pressure decline increases cardiovascular risk independent of blood pressure level—the “normotensive non-dipper” paradox. Chronobiol Int 30:87–98
Investigators A-H, Roush GC, Fagard RH, Salles GF, Pierdomenico SD, Reboldi G, Verdecchia P, Eguchi K, Kario K, Hoshide S, Polonia J, de la Sierra A, Hermida RC, Dolan E, Zamalloa H (2014) Prognostic impact from clinic, daytime, and night-time systolic blood pressure in nine cohorts of 13,844 patients with hypertension. J Hypert 32:2332–2340
Jennum P, Ibsen R, Knudsen S, Kjellberg J (2013) Comorbidity and mortality of narcolepsy: a controlled retro- and prospective national study. Sleep 36:835–840
Kayaba Y, Nakamura A, Kasuya Y, Ohuchi T, Yanagisawa M, Komuro I, Fukuda Y, Kuwaki T (2003) Attenuated defense response and low basal blood pressure in orexin knockout mice. Am J Physiol Regul Integr Comp Physiol 285:R581–R593
Kiyashchenko LI, Mileykovskiy BY, Maidment N, Lam HA, Wu MF, John J, Peever J, Siegel JM (2002) Release of hypocretin (orexin) during waking and sleep states. J Neurosci 22:5282–5286
Kovalska P, Kemlink D, Nevsimalova S, Maurovich Horvat E, Jarolimova E, Topinkova E, Sonka K (2016) Narcolepsy with cataplexy in patients aged over 60 years: a case-control study. Sleep Med 26:79–84
Kukkonen JP (2013) Physiology of the orexinergic/hypocretinergic system: a revisit in 2012. Am J Physiol Cell Physiol 304:C2–C32
Kuwaki T (2015) Thermoregulation under pressure: a role for orexin neurons. Temperature 2:379–391
Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E (1999) The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98:365–376
Lo Martire V, Silvani A, Bastianini S, Berteotti C, Zoccoli G (2012) Effects of ambient temperature on sleep and cardiovascular regulation in mice: the role of hypocretin/orexin neurons. PLoS One 7:e47032
Longstreth WT Jr, Koepsell TD, Ton TG, Hendrickson AF, van Belle G (2007) The epidemiology of narcolepsy. Sleep 30:13–26
Martelli D, Silvani A, McAllen RM, May CN, Ramchandra R (2014) The low frequency power of heart rate variability is neither a measure of cardiac sympathetic tone nor of baroreflex sensitivity. Am J Physiol Heart Circ Physiol 307:H1005–H1012
American Academy of Sleep Medicine (2014) International classification of sleep disorders. American Academy of Sleep Medicine, Darien
Ohayon MM (2013) Narcolepsy is complicated by high medical and psychiatric comorbidities: a comparison with the general population. Sleep Med 14:488–492
Ohayon MM, Black J, Lai C, Eller M, Guinta D, Bhattacharyya A (2014) Increased mortality in narcolepsy. Sleep 37:439–444
Orphanet (2016) List of rare diseases and synonyms listed in alphabetical order. http://www.orpha.net/orphacom/cahiers/docs/GB/List_of_rare_diseases_in_alphabetical_order.pdf. Accessed Dec 2016
Perez MV, Pavlovic A, Shang C, Wheeler MT, Miller CL, Liu J, Dewey FE, Pan S, Thanaporn PK, Absher D, Brandimarto J, Salisbury H, Chan K, Mukherjee R, Konadhode RP, Myers RM, Sedehi D, Scammell TE, Quertermous T, Cappola T, Ashley EA (2015) Systems genomics identifies a key role for hypocretin/orexin receptor-2 in human heart failure. J Am Coll Cardiol 66:2522–2533
Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, Nishino S, Mignot E (2000) A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med 6:991–997
Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS (1998) Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18:9996–10015
Plazzi G, Ferri R, Antelmi E, Bayard S, Franceschini C, Cosentino FI, Abril B, Spruyt K, Provini F, Montagna P, Dauvilliers Y (2010) Restless legs syndrome is frequent in narcolepsy with cataplexy patients. Sleep 33:689–694
Plazzi G, Moghadam KK, Maggi LS, Donadio V, Vetrugno R, Liguori R, Zoccoli G, Poli F, Pizza F, Pagotto U, Ferri R (2011) Autonomic disturbances in narcolepsy. Sleep Med Rev 15:187–196
Ponziani V, Gennari M, Pizza F, Balsamo A, Bernardi F, Plazzi G (2016) Growing up with type 1 narcolepsy: its anthropometric and endocrine features. J Clin Sleep Med 12:1649–1657
Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92:573–585
Samson WK, Gosnell B, Chang JK, Resch ZT, Murphy TC (1999) Cardiovascular regulatory actions of the hypocretins in brain. Brain Res 831:248–253
Sansa G, Iranzo A, Santamaria J (2010) Obstructive sleep apnea in narcolepsy. Sleep Med 11:93–95
Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE (2010) Sleep state switching. Neuron 68:1023–1042
Schwimmer H, Stauss HM, Abboud F, Nishino S, Mignot E, Zeitzer JM (2010) Effects of sleep on the cardiovascular and thermoregulatory systems: a possible role for hypocretins. J Appl Physiol 109:1053–1063
Shahid IZ, Rahman AA, Pilowsky PM (2012) Orexin A in rat rostral ventrolateral medulla is pressor, sympatho-excitatory, increases barosensitivity and attenuates the somato-sympathetic reflex. Br J Pharmacol 165:2292–2303
Shih CD, Chuang YC (2007) Nitric oxide and GABA mediate bi-directional cardiovascular effects of orexin in the nucleus tractus solitarii of rats. Neuroscience 149:625–635
Shirasaka T, Nakazato M, Matsukura S, Takasaki M, Kannan H (1999) Sympathetic and cardiovascular actions of orexins in conscious rats. Am J Physioll Regul Integr Comp Physiol 277:R1780–R1785
Sieminski M, Partinen M (2016) “Non-dipping” is equally frequent in narcoleptic patients and in patients with insomnia. Sleep Biol Rhythms 14:31–36
Silvani A (2008) Physiological sleep-dependent changes in arterial blood pressure: central autonomic commands and baroreflex control. Clin Exp Pharmacol Physiol 35:987–994
Silvani A (2017) Orexins and the cardiovascular events of awakening. Temperature 4:128–140. doi:10.1080/23328940.2017.1295128 (In press)
Silvani A, Bastianini S, Berteotti C, Cenacchi G, Leone O, Lo Martire V, Papa V, Zoccoli G (2014) Sleep and cardiovascular phenotype in middle-aged hypocretin-deficient narcoleptic mice. J Sleep Res 23:98–106
Silvani A, Bastianini S, Berteotti C, Lo Martire V, Zoccoli G (2012) Control of cardiovascular variability during undisturbed wake-sleep behavior in hypocretin-deficient mice. Am J Physiol Regul Integr Comp Physiol 302:R958–R964
Silvani A, Calandra-Buonaura G, Dampney RA, Cortelli P (2016) Brain–heart interactions: physiology and clinical implications. Phil Trans A Math Phys Eng Sci 374:20150181
Silvani A, Calandra-Buonaura G, Johnson BD, van Helmond N, Barletta G, Cecere AG, Joyner MJ, Cortelli P (2017) Physiological mechanisms mediating the coupling between heart period and arterial pressure in response to postural changes in humans. Front Physiol 8:163
Silvani A, Dampney RA (2013) Central control of cardiovascular function during sleep. Am J Physiol Heart Circul Physiol 305:H1683–H1692
Silvani A, Grimaldi D, Barletta G, Bastianini S, Vandi S, Pierangeli G, Plazzi G, Cortelli P (2013) Cardiovascular variability as a function of sleep-wake behaviour in narcolepsy with cataplexy. J Sleep Res 22:178–184
Sorensen GL, Knudsen S, Petersen ER, Kempfner J, Gammeltoft S, Sorensen HB, Jennum P (2013) Attenuated heart rate response is associated with hypocretin deficiency in patients with narcolepsy. Sleep 36:91–98
Takahashi K, Lin JS, Sakai K (2008) Neuronal activity of orexin and non-orexin waking-active neurons during wake-sleep states in the mouse. Neuroscience 153:860–870
Trenkwalder C, Allen R, Hogl B, Paulus W, Winkelmann J (2016) Restless legs syndrome associated with major diseases: a systematic review and new concept. Neurology 86:1336–1343
van der Meijden WP, Fronczek R, Reijntjes RH, Corssmit EP, Biermasz NR, Lammers GJ, van Dijk JG, Thijs RD (2015) Time- and state-dependent analysis of autonomic control in narcolepsy: higher heart rate with normal heart rate variability independent of sleep fragmentation. J Sleep Res 24:206–214
Wijnans L, Lecomte C, de Vries C, Weibel D, Sammon C, Hviid A, Svanstrom H, Molgaard-Nielsen D, Heijbel H, Dahlstrom LA, Hallgren J, Sparen P, Jennum P, Mosseveld M, Schuemie M, van der Maas N, Partinen M, Romio S, Trotta F, Santuccio C, Menna A, Plazzi G, Moghadam KK, Ferro S, Lammers GJ, Overeem S, Johansen K, Kramarz P, Bonhoeffer J, Sturkenboom MC (2013) The incidence of narcolepsy in Europe: before, during, and after the influenza A(H1N1)pdm09 pandemic and vaccination campaigns. Vaccine 31:1246–1254
Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, Leonard CS, Richardson JA, Hammer RE, Yanagisawa M (2003) Distinct narcolepsy syndromes in orexin receptor-2 and orexin null mice: molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron 38:715–730
Winkelman JW, Blackwell T, Stone K, Ancoli-Israel S, Redline S (2017) Associations of incident cardiovascular events with restless legs syndrome and periodic leg movements of sleep in older men, for the outcomes of sleep disorders in older men study (MrOS Sleep Study). Sleep 40(4). doi:10.1093/sleep/zsx023
Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, Mignot E, Nishino S (2001) Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci 14:1075–1081
Zhang W, Sakurai T, Fukuda Y, Kuwaki T (2006) Orexin neuron-mediated skeletal muscle vasodilation and shift of baroreflex during defense response in mice. Am J Physiol Regul Integr Comp Physiol 290:R1654–R1663
Zhong X, Hilton HJ, Gates GJ, Jelic S, Stern Y, Bartels MN, Demeersman RE, Basner RC (2005) Increased sympathetic and decreased parasympathetic cardiovascular modulation in normal humans with acute sleep deprivation. J Appl Physiol 98:2024–2032
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all the authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Berteotti, C., Silvani, A. The link between narcolepsy and autonomic cardiovascular dysfunction: a translational perspective. Clin Auton Res 28, 545–555 (2018). https://doi.org/10.1007/s10286-017-0473-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10286-017-0473-z