
Abstract
This study evaluated the effect of the combination of two dimethacrylate-based monomers [bisphenol A diglycidyl dimethacrylate (BisGMA) or bisphenol A ethoxylated dimethacrylate (BisEMA)] with diluents either derived from ethylene glycol dimethacrylate (ethylene glycol dimethacrylate, diethylene glycol dimethacrylate, triethylene glycol dimethacrylate, tetraethylene glycol dimethacrylate) or 1,10-decanediol dimethacrylate (D3MA) on network characteristics and mechanical properties of neat resin and composite materials. The degree of conversion, maximum rate of polymerization and water sorption/solubility of unfilled resins and the flexural strength and microhardness of composites (after 24 h storage in water and 3 months storage in a 75 vol% ethanol aqueous solution) were evaluated. Data were analyzed with two-way ANOVA and Tukey’s test (α = 0.05). The higher conversion and lower water sorption presented by BisEMA co-polymers resulted in greater resistance to degradation in ethanol compared with BisGMA-based materials. In general, conversion and mechanical properties were optimized with the use of long-chain dimethacrylate derivatives of ethylene glycol. D3MA rendered more hydrophobic materials, but with relatively low conversion and mechanical properties.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A high degree of conversion of the organic matrix is often related to superior mechanical properties in composite restorative materials, decreasing the risk of fracture and wear while in function [1, 2]. No less important, conversion is also related to decreased sorption of fluids and solubility of resin composites in the oral environment, which is crucial for a long-lasting clinical performance [3].
In dimethacrylate systems, such as the ones commonly used in dental composites, the viscosity of the non-polymerized material plays a major role in conversion. By combining base monomers and diluents, conversion kinetic parameters can be manipulated to maximize conversion prior to gelation, which in turn can potentially lead to higher limiting conversions [4–6]. Bisphenol A diglycidyl dimethacrylate (BisGMA), introduced in the early sixties [7], remains the most largely used monomer in dental composite formulations. Due to the presence of the bisphenol A core, which decreases the degrees of freedom for rotation around the bonds, and strong hydrogen bonding interactions given by the hydroxyl groups, this monomer presents extremely high viscosity at room temperature [8]. The addition of 25 to 50 % triethylene glycol dimethacrylate (TEGDMA) as a diluent results in decreased viscosity and improves handling characteristics, as well as allows for more expressive filler loadings [1, 9–12]. Another important feature is that the flexible TEGDMA improves conversion [11–14] and crosslinking [11, 15], resulting in fewer leachable components [16]. However, both these monomers are relatively hydrophilic [17], as for any molecule bearing ether bonds [18], which is a concern in terms of water sorption leading to long-term degradation [19].
In monomers derived from ethylene glycol, the number of ethylene glycol units influences the molecule’s hydrophilicity [20, 21]. For example, it has been shown that TEGDMA co-polymerized to BisGMA has resulted in greater compression strength and microhardness initially, but the use of a shorter derivate such as ethylene glycol dimethacrylate (EGDMA) led to greater polymer stability after water aging [19]. In similar fashion, tetraethylene glycol dimethacrylate (TETGDMA) is more hydrophilic than TEGDMA, but not to the point of negatively affecting fracture toughness or solubility resistance [22]. Other diluents are also used in dental resin composites, such as 1,10 decanediol dimethacrylate (D3MA) [23]. On the one hand, the absence of ethylene glycol units renders this molecule less hydrophilic, since it has fewer polar groups [24], but on the other hand, the longer spacer between methacrylates makes it more flexible and decreases the elastic modulus of the resulting polymer [25].
Bisphenol A ethoxylated dimethacrylate (BisEMA), the ethoxylated version of BisGMA, is another example of monomer used in dental resin composites. The molecular structure of such monomer is almost the same of BisGMA monomer, except for the absence of hydroxyl groups. It shows higher molecular weight than ethylene glycol derivates and intrinsically low viscosity, due to the absence of hydroxyl groups that form hydrogen bondings. The lower viscosity reduces the maximum rate of polymerization and enhances the final conversion achieved by the polymer, compared to neat BisGMA [6, 15]. However, a previous study noticed an increase in the viscosity when TEGDMA was partially or totally replaced by BisEMA as diluent when copolymerized with BisGMA base monomer, which was associated with decreases in conversion and flexural strength of the polymer [26].
Apart from the use of diluents, the incorporation of alternative base monomers is another way to tailor the system’s viscosity. For example, urethanes have been extensively investigated and used commercially [27, 28]. On the other hand, BisGMA analogs, such as BisEMA, have not been extensively also investigated as a base monomer in dental composites [29, 30]. The total replacement of BisGMA by BisEMA when associated with TEGDMA increased conversion, but mechanical strength remained similar [29]. Compared with BisGMA, BisEMA is less hydrophilic (due to the absence of hydroxyl groups) and establishes weaker hydrogen bond interactions with water molecules [23], which should promote a higher resistance to degradation in the long term. A recent study mentioned the necessity of further clarification regarding the influence of BisEMA as base monomer on conversion and sorption/solubility, since previous researches compared only commercial composites or unfilled resins [30].
New monomers have been used as alternative to dimethacrylate systems in organic matrix, such as dimer acid-based dimethacrylate, tricyclodecane-urethane and monomers that polymerize by ring-opening epoxy chemistry [12, 31]. Though some non-dimethacrylate systems have already been released in the market, the previously cited dimethacrylate monomers still represent the most commonly used in dental materials [12, 32]. When commercial resins containing dimer acid-based dimethacrylate and tricyclodecane-urethane monomers were compared to other commercial with usual dimethacrylate systems, they did not present better mechanical performance after aging in different solutions [31]. Even with respect to the common dimethacrylates, there are some combinations of monomers that still need to be studied regarding their physicochemical properties after storage.
The objective of this study was to evaluate the influence of different diluent monomers (ethylene glycol derivates and D3MA) in co-polymerizations with BisGMA or BisEMA, the last two used as base monomers, on the kinetics of conversion and water sorption/solubility of unfilled resins and on the mechanical properties before and after aging (ethanol 75 %) of composite materials. The tested hypothesis were the following: (1) BisEMA will present lower maximum polymerization rate and water sorption/solubility and higher conversion and mechanical properties before and after aging in ethanol; (2) ethylene glycol derivatives with higher molecular weight (TETGDMA and TEGDMA) will show higher conversion, maximum polymerization rate, water sorption/solubility and mechanical properties before and after aging than the lower molecular weight derivatives (DEGDMA and EGDMA); (3) D3MA will show conversion, maximum polymerization rate and initial mechanical properties similar to TEGDMA and TETGDMA (due to similar molecular weights) and greater than DEGDMA and EGDMA; water sorption/solubility will be lower and mechanical properties after aging will be greater than all ethylene glycol derivatives.
Materials and methods
Bisphenol A diglycidyl dimethacrylate (Evonik Industries, Essen, Germany) (Mw = 512 g/mol) and BisEMA (Esstech Inc., Essington, PA, USA) (Mw = 540 g/mol) were used as the base monomers and co-polymerized with five different diluents: ethylene glycol dimethacrylate (EGDMA; Mw = 198 g/mol), diethylene glycol dimethacrylate (DEGDMA; Mw = 242 g/mol), triethylene glycol dimethacrylate (TEGDMA; Mw = 286 g/mol) (all from Cognis Performance Chemicals, UK), tetraethylene glycol dimethacrylate (TETGDMA; Mw = 330 g/mol) (Evonik Industries, Essen, Germany) and 1,10-decanediol dimethacrylate (D3MA; Mw = 310 g/mol) (Cognis Performance Chemicals, UK). In all materials, the base monomer corresponded to 75 wt% of the organic matrix formulations. For the composite materials, 71 wt% barium aluminum silicate glass (Schott AG, Mainz, Germany, average particle size = 0.7 μm) was added mechanically under vacuum (SpeedMixer, FlackTek, Inc., Landrum, USA). The particles were silanized with 3-methacryloxypropyltrimethoxy silane (Evonik Industries, Essen, Germany). To render the materials photopolymerizable, 0.5 wt% camphorquinone (Esstech Inc., Essington, PA, USA) and 1 wt% dimethylamino dimethacrylate (Sigma-Aldrich, Steinheim, Germany) were added.
Experimental
Degree of conversion and maximum rate of polymerization
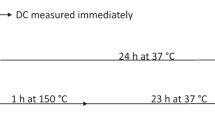
The degree of conversion and maximum rate of polymerization were determined by near infrared spectrometry (Nicolet 6700, Madison, WI, USA) configured with a white light source, extended KBr beam splitter and MCT/A detector. Spectra (n = 3) were acquired at a resolution of 4 cm−1 with 2 scans/s. Specimens (7-mm diameter × 1-mm thick) were photoactivated (16 J/cm2) for 40 s with an irradiance of 400 mW/cm2 (VIP Junior, Bisco Inc, Schaumburg, IL, USA). Before the photoactivation, the first four spectra were used to obtain data related to the non-polymerized material. From the start of the activation, the conversion was followed for 15 min. After 24-h dry storage at 37 °C, a new conversion measurement was taken. The degree of conversion was calculated in function of the area of vinyl overtone peak (6,165 cm−1) [33] obtained before polymerization and after 24 h. The rate of polymerization was calculated as the first derivative of the conversion × time curve, and the maximum value (RPmax) was recorded.
Water sorption and solubility
The water sorption and solubility was carried out according to ISO 4049 [34]. Disc-shaped specimens, 15 mm in diameter and 1 mm in thickness, were fabricated using a metallic mold. Materials (n = 5) were polymerized on both sides through a mylar strip at 16 J/cm2 (using the same parameters described for the conversion test), with five overlapping exposures of the light-cure tip to cover all the mold extension. Specimens were stored in a desiccator under vacuum at 37 °C until a stable mass reading was obtained. This was named m 1. The disc dimensions were measured with a digital caliper with a precision of 0.01 mm (Digimatic Caliper CD-6″OS, Mitutoyo, Japan), and then the volume (V) was calculated. Each specimen was then immersed in 20 mL distilled water at 37 °C. Subsequent mass measurements were taken at 1 h and at every 24 h in specimens blotted dry, until a stable mass reading (m 2) was obtained (approximately 40 days). The discs were then stored dry in the desiccator at 37 °C until a stable mass reading was obtained (m 3). Water sorption (W sp) and solubility (W sl) in μg/mm3 were calculated according to the Eqs. (1) and (2):
Flexural strength
Bar-shaped specimens (n = 20, 10 × 2 × 1 mm) were fabricated by placing the composite in a stainless steel split mold, sandwiched between glass slides. Materials were photopolymerized at 16 J/cm2 (using the same parameters described for the conversion test). Specimens’ dimensions were obtained with a digital caliper with 0.01-mm precision. Then, 10 specimens per group were stored in distilled water at 37 °C for 24 h, and the remaining specimens were stored in a 75 % solution of ethanol in distilled water. The storage solution was changed every week for 3 months, before the specimens were tested in 3-point bending, which was carried out in an universal testing machine (model 5565, Instron, Canton, MA, USA) with 8-mm span between supports and crosshead speed of 0.5 mm/min. Flexural strength was calculated with the Eq. (3), where P is the load at fracture (in N), L is the distance between the supports (8 mm), B is the width, and H is the height of the specimen (both in mm).
The fractured specimens were immersed in an ultrasonic cleaning bath (model T14, Thornton Inpec Eletronic Ltd., Vinhedo, SP, Brazil) with distilled water for 15 min, dried and sputter-coated with platinum (model MED 020, Bal-tec Coating System, Balzers, Leichtenstein). The fracture surfaces were then analyzed by SEM (Stereoscan 400, Cambridge, UK) up to 5,000× magnification.
Knoop microhardness
The microhardness test (n = 3) was performed in one of the fragments from the flexural strength test in a microhardness tester (model HMV-2T, Shimadzu Corporation, Kyoto, Japan). Six indentations were made in the non-irradiated surface of the specimen, under 25 g of load applied for 10 s. The Knoop hardness number (KHN) value was the average of these six indentations.
Statistical analysis
Conversion, water sorption/solubility, flexural strength and microhardness data were analyzed with two-way ANOVA and Tukey’s test. Student’s t test was used to compare values of flexural strength and microhardness before and after aging. The global level of significance was set to 5 %.
Results
Results of degree of conversion, maximum rate of polymerization, water sorption and solubility for the unfilled materials are shown in Table 1.
Degree of conversion and maximum rate of polymerization
For degree of conversion, the interaction was not statistically significant (p = 0.099). Materials containing BisEMA showed statistically higher conversion than BisGMA-based materials (pooled averages, respectively, 60.9 ± 3.2 and 54.7 ± 2.2 %, p < 0.01). Tetraethylene glycol dimethacrylate (60.3 ± 3.7 %a) and TEGDMA (59.7 ± 4.1 %ab) showed higher conversion than DEGDMA (57.5 ± 3.6 %c) and EGDMA (53.2 ± 2.6 %d, p < 0.01). 1,10 Decanediol dimethacrylate (58.5 ± 3.6 %bc) reached conversion similar to TEGDMA and DEGDMA, but lower than TETGDMA. Only the factor base monomer showed a statistically significant effect on the maximum rate of polymerization. Bisphenol A diglycidyl dimethacrylate-based materials showed higher maximum rates of polymerization compared to BisEMA (6.2 ± 0.8 and 3.9 ± 0.6 % s−1, respectively, p < 0.001).
Water sorption and solubility
When co-polymerized with BisGMA, TETGDMA and TEGDMA showed greater water sorption than DEGDMA, while EGDMA was higher than D3MA. For BisEMA-based materials, TETGDMA presented greater sorption than DEGDMA and EGDMA, while TEGDMA and D3MA were statistically similar to all other diluents (p < 0.01). The factors base monomer and diluent and the interaction between them did not show significant effects on solubility (p > 0.05).
Flexural strength
For flexural strength at 24 h, the interaction (p = 0.245) and the base monomer (p = 0.102) were not statistically significant. Formulations containing TEGDMA presented initial flexural strength values statistically higher than those containing D3MA (143.6 ± 17.4 MPaa and 128.5 ± 15.3 MPab, respectively) (p < 0.05) (Fig. 1). Ethylene glycol (134.9 ± 20.4 MPaab), DEGDMA (142.7 ± 23.6 MPaab) and TETGDMA (130.5 ± 17.6 MPaab) behaved statistically similar to both. Storage in ethanol solution significantly reduced flexural strength (p < 0.001), between 82 and 91 % for BisGMA copolymers and between 68 and 76 % for BisEMA, respectively. After 3 months, BisEMA containing formulations presented higher flexural strength than BisGMA (36.1 ± 9.6 and 16.9 ± 6.8 MPa, respectively, p < 0.01). Tetraethylene glycol dimethacrylate (32.4 ± 12.1 MPaa) presented higher flexural strength than EGDMA (22.8 ± 12.4 MPab) and D3MA (22.8 ± 12.3 MPab). DEGDMA (26.0 ± 14.6 MPaab) and TEGDMA (28.5 ± 10.4 MPaab) showed strength values after 3 months similar to the other diluents (p < 0.01).

Flexural strength (a) and Knoop microhardness (b) of the resin composites before and after 3 months storage in ethanol
The fracture surface in all groups revealed a brittle mode of failure, both before and after immersion in ethanol (Fig. 2). Hackle lines point to the origin of fracture in minor porosities within the material (Fig. 2a). The crack seemed to propagate through the organic matrix, as no exposed filler particles were observed (Fig. 2b).

SEM micrographs of composite fractured surfaces. a Surface under ×250 magnification showing the fracture origin (asterisk); the arrows indicate the semi-ellipse suggestive of subcritical crack growth. b Surface under ×5,000 magnification showing filler particles covered by matrix
There was no difference in the overall shape of the load × displacement curves for the different diluent monomers within the same base monomer (overall shapes are shown in Fig. 3). It can be noticed a considerable reduction in the elastic region in the load vs. displacement curves associated with elution of monomer from the polymer matrix after 3 months, which was very similar among all the groups (Table 1).

Overall shapes of load × displacement curves of BisGMA- and BisEMA-based resins in flexure after 24 h and 3 months (3 m)
Knoop microhardness
For KHN after 24 h, the interaction was statistically significant (p < 0.001). Formulations containing BisEMA and EGDMA derivatives showed statistically similar averages, all higher than D3MA (Fig. 1). Formulations containing BisGMA, TEGDMA and TETGDMA showed higher KHN than D3MA, while EGDMA and DEGDMA were similar to both. After 3 months in ethanol, formulations containing BisGMA showed reductions between 41 and 50 %. For the BisEMA-based formulations, reductions ranged from 10 to 37 %. After 3 months, formulations containing BisEMA showed higher KHN than the BisGMA-based ones (18.1 ± 1.8 and 15.3 ± 2.5, respectively, p < 0.01). Ethylene glycol (18.4 ± 2.7a) presented higher KHN compared to D3MA (14.7 ± 2.0b). DEGDMA (16.5 ± 2.6ab), TEGDMA (16.1 ± 2.4ab) and TETGDMA (17.8 ± 2.2ab) showed average KHN similar to the other diluents (p < 0.05).
Discussion
Although new monomers have been developed, co-polymers of dimethacrylates are the most commonly used in dental resin composites [8]. In the present study, the usual commercial proportion between base and diluent monomers of 75:25 by weight was used, since it allows for high filler loadings to be achieved [15]. It is noteworthy that, due to differences in molecular weights among the monomers, the molar ratio of base/diluent was slightly different (0.9 for EGDMA; 0.7 for DEGDMA; 0.6 for TEGDMA; 0.5 for TETGDMA; and 0.5 for D3MA). These values were very similar for BisGMA and BisEMA, due to their similar molecular weights (512 and 540 g/mol, respectively).
Degree of conversion and maximum rate of polymerization
Hypothesis 1 was accepted, as conversion achieved by BisEMA was greater than BisGMA. This finding is in accordance with a previous study that found higher conversion when BisGMA was totally or partially replaced by BisEMA as base monomer with all irradiation times used [30]. Compared to BisGMA, BisEMA longer ethylene glycol spacer increases flexibility, while its lower initial viscosity, granted by the weaker intermolecular interaction, facilitates diffusion prior to gelation, ultimately leading to a higher limiting conversion [6, 15]. As far as the diluents are concerned, hypothesis 2 was also accepted. Tetraethylene glycol dimethacrylate and TEGDMA presented higher conversion compared to DEGDMA and EGDMA, likely due to the greater reactivity associated with longer spacers (i.e., higher number of ethylene glycol units) between methacrylate groups [35]. Indeed, other studies have demonstrated that the reactivity decreases as the number of ethylene glycol units decreases [36] due to a reduced flexibility [37]. When comparing TETGDMA and D3MA, which have similar distances between methacrylate groups, the role of flexibility is highlighted, since the presence of ethylene glycol units in TETGDMA provides additional degrees of freedom to the molecule. This leads to the partial rejection of hypothesis 3.
The strong intermolecular interactions in BisGMA and its consequent high monomeric viscosity favor propagation at earlier stages of conversion compared to the less hindered BisEMA [6, 14, 30, 32]. That way, gelation in BisGMA is achieved earlier, which causes the maximum rates of polymerization to be higher and the limiting values of conversion lower, in comparison with BisEMA-based materials. One explanation for the lack of influence of diluent monomers on polymerization rate is that the initial viscosity is virtually the same in all materials (within the same base monomer), since the intermolecular interactions are similar (all diluent monomers are only hydrogen bond acceptors, not donors). Therefore, in regard to maximum rate of polymerization, hypothesis 1 was accepted and hypotheses 2 and 3 were rejected.
Water sorption and solubility
Higher hydrophilicity and lower degree of conversion were expected to render polymers with greater water sorption [6, 21, 23, 24, 38, 39], as observed in this work when comparing BisGMA to BisEMA-based polymers. A recent study observed lower sorption with partial or total replacement of BisGMA by BisEMA [30]. Hydrogen bonds between water molecules and BisGMA hydroxyl groups is three orders of magnitude stronger than the ones that can be established with ether groups, present both in BisGMA and in BisEMA [3, 40]. The free volume which is a result of molecule packing in the polymer also influences water uptake [39, 41]. In spite of the similar intermolecular interactions developed between diluents and base monomers within the same base monomer, the differences in polymer packing given by the ethylene glycol derivatives (and their different spacer sizes between methacrylates) and the D3MA were able to produce differences in water sorption. It is likely that EGDMA and DEGDMA have produced tighter networks compared to TEGDMA and TETGDMA, since their smaller chains produce shorter crosslinks [42]. Another factor that needs to be considered is the fact that lower molecular weight monomers were actually in a higher molar concentration. In other words, for EGDMA and DEGDMA, the crosslinker concentration was even higher and that allied to the shorter chains, probably produced a denser network and led to less water sorption [39]. 1,10 Decanediol dimethacrylate does not present hydrogen bonding acceptor sites in its backbone, providing a hydrophobic character, and the water sorption, as a consequence, was lower [23]. In the BisEMA formulations, the water sorption values were closer to each other, probably because the base monomer has a greater hydrophobic character. In those cases, the higher sorption observed for TETGDMA when compared to EGDMA and DEGDMA was also a function of the longer crosslinks formed in the network. This leads to the acceptance of hypothesis 1 and partial rejection of hypotheses 2 and 3.
In regard to solubility, all groups showed statistically similar results. Such finding suggests that conversion and amount of water absorbed into the network are not the only factors affecting solubility, and hydrophilic character of leachable present in the structure (monomers and oligomers) also plays a role [6]. Only 2–8 % of the remaining double bonds correspond to free residual monomer [39, 43, 44], meaning that the remainder of the unreacted double bonds remains pendant from the network structure. The pendant double bonds contribute to water sorption, but not to leachable species [6].
When in contact with the network, the solvent establishes attraction forces with polymeric components, breaking the secondary bonds between chains and consequent plasticization of the material. Surface properties such as hardness tend to be affected in earlier stages than bulk properties, which are affected more slowly [39]. Although both base monomers have presented similar solubilities, BisGMA tends to suffer more degradation with decrease in mechanical properties in long term than BisEMA due to the higher water sorption.
Flexural strength
Bisphenol A ethoxylated dimethacrylate and BisGMA presented similar early flexural strength, as observed in the literature [29], in spite of the higher conversion presented by the former. It is a clear indication of the role of hydrogen bonding on the mechanical properties of the resulting network, since they work as non-covalent crosslinks in the polymer [8, 45]. However, hydrogen bonds are exactly the secondary bonds that are prone to be attacked by protic solvents, such as water and ethanol, then leading to a greater decrease in flexural strength after storage in 75 % ethanol solution for 3 months for the BisGMA-based materials, as expected [39, 41]. For BisEMA, the greater resistance to degradation by organic solvents was determined not only by the more hydrophobic character, but also by the higher degree of conversion. As for the diluents, the small differences in conversion did not reflect in the early mechanical properties. The lower conversion presented by EGDMA and DEGDMA seems to have been compensated by the tighter network given by the shorter crosslinks and also by the relatively greater molar concentration of diluents, compared to TEGDMA and D3MA. Comparing the last two monomers, the more flexible D3MA led to lower initial flexural strength compared to TEGDMA, as expected [25], in spite of the higher conversion.
The analysis of the fractured surfaces under SEM shows hackle lines pointing to the origin of fracture in minor porosities within the material. Also, the load × displacement curves showed that almost all the deformation occurred in the elastic regime. These two factors combined indicate that there was slow crack propagation and absence of plastic deformation, suggestive of a subcritical crack growth behavior [46].
Knoop microhardness
The microhardness values followed the same trends observed for flexural strength: within the same diluent, BisGMA and BisEMA presented similar KHN values prior to storage in ethanol. The only exception was BisEMA co-polymerized to D3MA, showing the lowest mean value among all formulations, likely due to the high flexibility of the diluent, allied to the lack of strong intermolecular interactions given by the base monomer. After storage, BisEMA formulations have again proven better able to resist degradation. In this case, no difference was observed among diluents, likely because the surface mechanical properties are more sensitive to the plasticization effects of the solvent [41]. In regard to initial flexural strength and microhardness, all hypotheses were rejected, while for the results after aging, hypothesis 1 was accepted and hypotheses 2 and 3 were rejected.
According to previous studies, when BisGMA was used as base monomer, the replacement of TEGDMA by BisEMA as diluent was not advantageous for degree of conversion nor flexural strength of the resultant polymer [26], and when BisEMA replaced BisGMA as base monomer when used with TEGDMA, there was no improvement in mechanical properties, which remained similar, in spite of a higher conversion [29]. Nevertheless, the findings of the present study indicate that, although the total replacement of BisGMA by BisEMA as base monomer did not improve immediate mechanical properties, it decreased water sorption and degradation after aging, which suggests a better performance in long-term use.
Conclusions
Due to its higher flexural strength and microhardness after aging and its lower water sorption, BisEMA appears to be a suitable alternative to BisGMA as base monomer in resin composites. DEGDMA, TEGDMA and TETGDMA showed a better compromise among conversion and water sorption compared to EGDMA, ultimately leading to better mechanical properties. 1,10 Decanediol dimethacrylate did not improve mechanical properties before or after storage in spite of is greater hydrophobicity in relation to the ethylene glycol derivatives.
References
Ferracane JL, Greener EH. The effect of resin formulation on the degree of conversion and mechanical properties of dental restorative resins. J Biomed Mater Res. 1986;20(1):121–31.
Bouschlicher MR, Rueggeberg FA, Wilson BM. Correlation of bottom-to-top surface microhardness and conversion ratios for a variety of resin composite compositions. Oper Dent. 2004;29(6):698–704.
Sideridou I, Tserki V, Papanastasiou G. Study of water sorption, solubility and modulus of elasticity of light-cured dimethacrylate-based dental resins. Biomaterials. 2003;24(4):655–65.
Odian G. Principles of polymerization. 3rd ed. New York: John Wiley & Sons; 1994.
Andrzejewska E. Photopolymerization kinetics of multifunctional monomers. Prog Polym Sci. 2001;26(4):605–65.
Gajewski VE, Pfeifer CS, Froes-Salgado NR, Boaro LC, Braga RR. Monomers used in resin composites: degree of conversion, mechanical properties and water sorption/solubility. Braz Dent J. 2012;23(5):508–14.
Bowen RL (1962) Inventor dental filling material comprising vinyl-silane treated fused silica and a binder consisting of the reaction product of bisphenol and glycidyl methacrylate. US Patent 3,066,112, 27 Nov 1962.
Stansbury JW. Dimethacrylate network formation and polymer property evolution as determined by the selection of monomers and curing conditions. Dent Mater. 2012;28(1):13–22.
Peutzfeldt A. Resin composites in dentistry: the monomer systems. Eur J Oral Sci. 1997;105(2):97–116.
Davy KW, Kalachandra S, Pandain MS, Braden M. Relationship between composite matrix molecular structure and properties. Biomaterials. 1998;19(22):2007–14.
Pfeifer CS, Shelton ZR, Braga RR, Windmoller D, Machado JC, Stansbury JW. Characterization of dimethacrylate polymeric networks: a study of the crosslinked structure formed by monomers used in dental composites. Eur Polymer J. 2011;47(2):162–70.
Cramer NB, Stansbury JW, Bowman CN. Recent advances and developments in composite dental restorative materials. J Dent Res. 2011;90(4):402–16.
Feilzer AJ, Dauvillier BS. Effect of TEGDMA/BisGMA ratio on stress development and viscoelastic properties of experimental two-paste composites. J Dent Res. 2003;82(10):824–8.
Lovell LG, Newman SM, Bowman CN. The effects of light intensity, temperature, and comonomer composition on the polymerization behavior of dimethacrylate dental resins. J Dent Res. 1999;78(8):1469–76.
Sideridou I, Tserki V, Papanastasiou G. Effect of chemical structure on degree of conversion in light-cured dimethacrylate-based dental resins. Biomaterials. 2002;23(8):1819–29.
Floyd CJ, Dickens SH. Network structure of Bis-GMA- and UDMA-based resin systems. Dent Mater. 2006;22(12):1143–9.
Atai M, Nekoomanesh M, Hashemi SA, Amani S. Physical and mechanical properties of an experimental dental composite based on a new monomer. Dent Mater. 2004;20(7):663–8.
Beatty MW, Swartz ML, Moore BK, Phillips RW, Roberts TA. Effect of crosslinking agent content, monomer functionality, and repeat unit chemistry on properties of unfilled resins. J Biomed Mater Res. 1993;27(3):403–13.
Krishnan VK, Manjusha K, Yamuna V. Effect of diluent upon the properties of a visible-light-cured dental composite. J Mater Sci Mater Med. 1997;8(11):703–6.
Arima T, Hamada T, McCabe JF. The effects of cross-linking agents on some properties of HEMA-based resins. J Dent Res. 1995;74(9):1597–601.
Park J, Eslick J, Ye Q, Misra A, Spencer P. The influence of chemical structure on the properties in methacrylate-based dentin adhesives. Dent Mater. 2011;27(11):1086–93.
Indrani DJ, Cook WD, Televantos F, Tyas MJ, Harcourt JK. Fracture toughness of water-aged resin composite restorative materials. Dent Mater. 1995;11(3):201–7.
Sideridou ID, Karabela MM, Vouvoudi E. Volumetric dimensional changes of dental light-cured dimethacrylate resins after sorption of water or ethanol. Dent Mater. 2008;24(8):1131–6.
Sideridou ID, Karabela MM. Sorption of water, ethanol or ethanol/water solutions by light-cured dental dimethacrylate resins. Dent Mater. 2011;27(10):1003–10.
Moszner N, Fischer UK, Angermann J, Rheinberger V. Bis-(acrylamide)s as new cross-linkers for resin-based composite restoratives. Dent Mater. 2006;22(12):1157–62.
Goncalves F, Kawano Y, Pfeifer C, Stansbury JW, Braga RR. Influence of BisGMA, TEGDMA, and BisEMA contents on viscosity, conversion, and flexural strength of experimental resins and composites. Eur J Oral Sci. 2009;117(4):442–6.
Charton C, Falk V, Marchal P, Pla F, Colon P. Influence of Tg, viscosity and chemical structure of monomers on shrinkage stress in light-cured dimethacrylate-based dental resins. Dent Mater. 2007;23(11):1447–59.
Ellakwa A, Cho N, Lee IB. The effect of resin matrix composition on the polymerization shrinkage and rheological properties of experimental dental composites. Dent Mater. 2007;23(10):1229–35.
Stansbury JW. Synthesis and evaluation of novel multifunctional oligomers for dentistry. J Dent Res. 1992;71(3):434–7.
Cornelio RB, Wikant A, Mjosund H, Kopperud HM, Haasum J, Gedde UW, et al. The influence of bis-EMA vs bis GMA on the degree of conversion and water susceptibility of experimental composite materials. Acta Odontol Scand. 2013 (in press).
Schmidt C, Ilie N. The effect of aging on the mechanical properties of nanohybrid composites based on new monomer formulations. Clin Oral Invest. 2013;17(1):251–7.
Leprince JG, Palin WM, Hadis MA, Devaux J, Leloup G. Progress in dimethacrylate-based dental composite technology and curing efficiency. Dent Mater. 2013;29(2):139–56.
Stansbury JW, Dickens SH. Determination of double bond conversion in dental resins by near infrared spectroscopy. Dent Mater. 2001;17(1):71–9.
ISO 4049:2000. Dentistry: polymer-based filling, restorative and luting materials. International Organization for Standardization, Geneva. 2000.
Moore JE. Photopolymerization of multifunctional acrylates and methacrylates. Am Chem Soc. 1976;36:747–53.
Anseth KS, Kline LM, Walker TA, Anderson KJ, Bowman CN. Reaction-kinetics and volume relaxation during polymerizations of multiethylene glycol dimethacrylates. Macromolecules. 1995;28(7):2491–9.
Ruyter IE, Svendsen SA. Remaining methacrylate groups in composite restorative materials. Acta Odontol Scand. 1978;36(2):75–82.
Wei YJ, Silikas N, Zhang ZT, Watts DC. Diffusion and concurrent solubility of self-adhering and new resin-matrix composites during water sorption/desorption cycles. Dent Mater. 2011;27(2):197–205.
Ferracane JL. Hygroscopic and hydrolytic effects in dental polymer networks. Dent Mater. 2006;22(3):211–22.
Van Krevelen D. Properties of polymers. 3rd ed. Amsterdam: Elsevier; 1990.
Ferracane JL. Is the wear of dental composites still a clinical concern? Is there still a need for in vitro wear simulating devices? Dent Mater. 2006;22(8):689–92.
Bland MH, Peppas NA. Photopolymerized multifunctional (meth)acrylates as model polymers for dental applications. Biomaterials. 1996;17(11):1109–14.
Ferracane JL. Elution of leachable components from composites. J Oral Rehabil. 1994;21(4):441–52.
Tanaka K, Taira M, Shintani H, Wakasa K, Yamaki M. Residual monomers (TEGDMA and Bis-GMA) of a set visible-light-cured dental composite resin when immersed in water. J Oral Rehabil. 1991;18(4):353–62.
Lemon MT, Jones MS, Stansbury JW. Hydrogen bonding interactions in methacrylate monomers and polymers. J Biomed Mater Res A. 2007;83(3):734–46.
Montes GG, Draughn RA. Slow crack propagation in composite restorative materials. J Biomed Mater Res. 1987;21(5):629–42.
Acknowledgments
The authors would like to express their gratitude to CAPES (Coordination for Enhancement of Higher Education Personnel) for the financial support (PNPD-CAPES 02436/09-4), to Antonio Carlos Lascala for the technical support and to FGM Produtos Odontológicos (Joinville, SC, Brazil) for preparing the experimental composites tested in this study.
Conflict of interest
The authors Nívea R. G. Fróes-Salgado, Vinícius Gajewski, Bárbara P. Ornaghi, Carmem S. Pfeifer, Marcia M. Meier, Tathy Aparecida Xavier and Roberto R. Braga declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Godoy Fróes-Salgado, N.R., Gajewski, V., Ornaghi, B.P. et al. Influence of the base and diluent monomer on network characteristics and mechanical properties of neat resin and composite materials. Odontology 103, 160–168 (2015). https://doi.org/10.1007/s10266-014-0153-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10266-014-0153-6