
Abstract
The group of diseases classified as seronegative spondyloarthritis or enthesoarthritis is characterized by typical osteoarticular and extra-articular manifestations. Diverse patterns of disease can affect different members of the same family and may show different features in the same patient, with clinical overlaps thwarting the differential diagnosis. An anatomo-pathological hallmark in enthesoarthritis is the inflammatory process in the synovio-entheseal sites. The inflammatory microenvironment of synovio-entheseal complex, named enthesitis, is characterized, after an initial inflammatory/erosive phase, by a subsequent phase of neobone apposition, which seems to progress independently from the previous erosive phase, suggesting that the physiopathogenetic mechanisms that underlay the two phases are driven by different pivots. The structural damage is characterized by excessive neobone formation, with the syndesmophyte as a typical lesion. The process underlying their formation is not fully understood, although there are many useful information to clarify the physiopathogenetic puzzle. The primum movens of the enthesitic process is the micro-trauma to which entheses are subject, especially in the lower limbs, for biomechanical reasons. The inflammatory process is facilitated by the sequential structure of the organ enthesis, constitutionally devoid of sub-enthesitic cortical bone and closely related to the underlying trabecular bone and the medullary vascular system. The reparative attempt from the vascular system, thanks to the activating action of certain loco-regional cytokines, such as TNF α, conditions the possible deposit in the enthesis of molecules derived from other organic sites and able, especially in HLA-B27+ subjects, to activate and self-renew an immune-mediated inflammatory process following the initial mechanical process.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
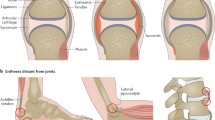
The seronegative spondyloarthritis or enthesoarthritis affects about 0.5 % of the population, and ankylosing spondylitis (AS), which together with reactive arthritis, psoriatic arthritis and enteropathic arthritis is part of it, affects 0.1 % of the population. There are three points to make about this group of diseases: (a) dissimilar aspects of the different phenotypic disease may occur in the same patient; (b) such clinical overlaps make difficult the differential diagnosis between a disease and the other; (c) spondyloarthritis belonging to different diseases can affect different members of the same family. The typical clinical extra-articular and osteoarticular manifestations of spondyloarthritis are reported in Table 1, and the anatomo-pathological hallmarks in enthesoarthritis are reported in Table 2. The characteristic of spondyloarthritis is inflammation of enthesis, a thin fibrocartilaginous area that joints the tendon/ligament or articular capsules to bone [1]. However, this inflammation has consequences not only on the enthesis per se. The concept of enthesis as an organ stems precisely from the consideration that a large anatomical area participates in the inflammatory process. In fact, the high mechanical stress that the insertional tendon, hinging point between hard and soft tissues, undergoes not only focuses on a small enthesis area but is distributed on a much larger area around it, with the aim to supply a biomechanical compensation. This area is determined by the periosteal and sesamoid fibrocartilage, synovial membrane, bursa, fat tissue, and sub-enthesitic spongy bone tissue. The archetypal enthesis organ is the Achilles tendon, a typical location involved in AS and in spondyloarthritis in general (Fig. 1). The Achilles tendon is inserted with a broad aponeurosis on the posterior surface of the calcaneus globally occupying its middle third and bottom and letting the upper third, the seat of the superior tuberosity. The latter, coming into contact with the tendon in the movements of dorsiflexion of the foot, plays a vital pulley role downloading the biomechanical stress on the tendon pre-enthesitic area. The same tendon, consequently to repeated contact with the superior tuberosity, is covered with fibrocartilage tissue on its deep surface, the so-called sesamoid fibrocartilage (Fig. 1), rich in proteoglicanic extracellular matrix and water in sufficient quantity and quality to ensure to the tendon the ability to withstand compressive forces. The greater tuberosity itself loses its fibrous periosteum in favor of a cartilage, the so-called periosteal fibrocartilage. Between the two fibrocartilages, in order to download further mechanical clutches, fits the retrocalcaneal bursa, provided inside with an additional cooling system of mechanical stress, which is the fatty tissue. Two other features, proprioceptive and immunological, are recognized in intrabursal adipose tissue. The latter is related to the wealth of macrophage cells [1].

Synovio-entheseal complex at the level of the Achilles tendon. The enthesis is a fibrocartilaginous structure and extends to sesamoid and periosteal fibrocartilage covering, respectively, the deep surface of the tendon and the superficial cortical bone at the level of the superior tuberosity. The micro-traumatic areas are localized in the areas of the enthesis and in the wall of the bursa. The blood vessels originate from the underlying bone at sites where there is no subchondral bone
The anatomical e functional concepts of “synovio-entheseal complex”
The synovio-entheseal complex identifies interdependence between two anatomical and functional structures of the organ enthesis, the synovial membrane and the enthesis itself. The enthesis is avascular, devoid of macrophages, dominated by extravascular matrix and prone to undergo microstructural damage caused by mechanical stress, whereas the synovial membrane is vascularized, rich in macrophages, consists mainly of loose connective and adipose tissue and is ready to generate inflammation. Therefore, the micro-damage manifesting itself on the enthesis goes with an inflamed synovium. In fact, in the Achilles tendon of AS patients, there are frequent problems such as bursitis or multifocal bone erosion of the superior tuberosity [2].
The fibrocartilages of enthesis organ are, as mentioned, avascular, but following their micro-damage, increasingly correlated with age, a reparative attempt occurs coming from the underlying bone associated with neoangiogenesis encroaching the enthesis and the periosteal fibrocartilage (Fig. 1). This process, facilitated by the limited presence of cortical bone underlying the enthesitic fibrocartilage, could transform entheses in seats of deposit of adjuvant molecules of bacterial origin from distant sites (intestine and other locations) with the consequences of immunological disturbance and subsequent local inflammation especially in the case of a concomitant genetic condition, such as the HLA-B27 positivity. The previously emphasized absence of sub-enthesitic cortical bone also facilitates the invasion on site of inflammatory cells coming from the bone marrow and the dissipation of enthesitic stress in cancellous bone. This latter aspect would explain why the osteitis is a clinical–radiological event often associated with the enthesitic process. Other vessels invading the tendon come from the adipose tissue and the peritendinous connective tissue [3].
It is possible that an innate immune response is activated at sites of damaged enthesis. In fact, a large number of damage-associated molecular patterns (DAMPs) have been identified in the enthesitic seats, which would activate the innate immune response [4]. The reaction to inflammation of the enthesis extends to the neighboring tissues, and among these, to the bone, being the osteitis adjacent to the synovial or enthesitic fibrocartilage a typical clinical and radiographic presentation of AS [3]. This framework is expressed exuberantly in some syndromes associated with seronegative spondyloarthritis, such as the SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, and osteitis) [5, 6].
The time evolution of exuberant and reparative attempts leads to the formation of enthesophytes originated by the superficial parts of the thin sub-enthesitic cortical layers with extension in the enthesitic regions (Fig. 2). Common elements in the formation of enthesophytes are also the absence in their context of trabecular bone and their development in areas lacking entirely, or nearly so, the thin layer of cortical bone as to express their reparative response coming from the subcortical trabecular bone. In such a place, cartilaginous islands can be traced between the trabecular bones as an expression of that reparative attempt, especially in the vicinity of horizontal cortical micro-cracks. Trabecular bone is, therefore, functionally integrated in the organ enthesis. This is demonstrated also by the thick trabecular spiculature, located in cancellous bone to a depth of 2–4 mm, whose directional trend follows that of the tendon to express a “deepening” of the enthesitic arm in the depth of cancellous bone. These aspects are clearly visible to a histological and radiological assessment and are mainly expressed in the seats of the enthesitic sites of the lower limbs where the load and entheses micro-damage are greater. This system of thickening and orientation of the sub-enthesitic bone trabeculae integrates with an underlying system of trabecular “hooking” characterized by a direction of the trabeculae perpendicular to that of the tendon, and with the function of stabilizing the enthesitic system. These anatomical and functional patterns, more frequent in old age, can actually affect any age and express the repair of micro-damages in enthesitic locations. This same mechanism, when located in the vertebrae, could lead to the formation of syndesmophytes in AS. In this case, the local enthesis micro-damage entails a reparative attempt of the inflammatory damage, expressed by the invasion of micro-vessels at the enthesis–bone interface. Secondly, the same vascular invasion could facilitate the “deposition” in the enthesitic seats of adjuvants molecules, of bacterial origin, in particular intestinal bacteria with the activation of loco-regional innate immunity [7]. An autoimmune process against local antigens “uncovered” by the micro-damage repeating over time could add especially in HLA-B27-positive subjects [8]. Therefore, in the pathogenesis of enthesoarthritis, multiple pathogenic mechanisms may converge, initially mechanical–inflammatory (non-immune) and then immune-mediated mechanisms of both innate and adaptive nature with the mediation of HLA class I [9].

Enthesis organ. From an anatomical point of view, there is not a real area of compact bone below the enthesitic fibrocartilage. The coating of cortical bone in these locations is extremely small ranging from 50 to 600 μm compared to nearly 3 mm elsewhere. In the line of demarcation of the cortical bone surface in many enthesitic locations bone presents breaks, micro-cracks or horizontal holes of 100–400 μm probably secondary to micro-damage of enthesitic fibrocartilage transmitted in adjacent structures, more frequent in old age and in the enthesis of the lower limbs. Moreover, in almost all enthesitic locations, there are micro-areas of complete absence of cortical bone so that the underlying spongy bone comes into direct contact with enthesitic fibrocartilage through the micro-cracks. Through this enthesis-trabecular bone contact trabecular bone tries to repair the micro-damage of fibrocartilaginous enthesis with invasion of blood vessels and connective tissue cells. In such locations signs of remodeling of both the thin cortical sub-enthesitic and underlying trabecular bone are evident. Signs of this remodeling are the presence of osteoid tissue, osteoclasts and osteoblasts, and immune cells and stem cells. The simultaneous presence, in the subtle subcortical enthesitic locations, of lamellar bone alternating with non-lamellar bone pinpoints the presence of reparative rearrangements occurring at different times
The inflammatory microenvironment of AS
Study models
An experimental model that renders what is recorded in humans in the course of AS is that of DBA1 mouse in which “spontaneous” arthritis is followed by “joint fusion.” In this model, the bone morphogenetic proteins (BMPs), belonging to the superfamily of TGF-β ligands, are able to modulate the initial stages of formation of osteophytes through activation of Wnt signaling [10, 11]. Moreover, in the same model, the inhibition of TNF-α has a small effect on the activation of SMAD protein 1/5 (mediators downstream of BMP signaling) and therefore is not able to alter the extension of new bone formation and bone ankylosis [12]. Thus, even the block of osteoclastic resorptive activity with zoledronic acid does not reduce the severity of bone ankylosis, suggesting that it is independent of the resorptive osteoclastic activity in AS [13].
In a second model, the hTNFtg mouse, in which there is a constitutional hyper-expression of TNF-α resulting in spontaneous development of arthritis, the block of the antagonist of Wnt signaling, DKK1 (Dickkopf 1, inhibitor of the osteoblastogenic function of Wnt signaling), determines osteophytic-like bone neoformation in sites where before there was an erosion. Therefore, the Wnt proteins have, in the reported animal model, a role in regulating the imbalance between neoformation and bone resorption in favor of the former [14, 15].
In the same mouse model, the blockade of osteoclast function by exploiting a possible defective expression of the transcription factor c-Fos [16, 17] or by treatment with bisphosphonates prevents erosion emphasizing the central role played by osteoclasts during the erosive phase of arthritis. Therefore, even blocking the main osteoclast differentiation factor receptor activator of nuclear factor-kB ligand (RANKL) protects from erosion in a mouse model of spontaneous arthritis [18].
The role of osteoclasts and osteoblasts
Osteoclast activation in inflammatory erosive sites of rheumatoid arthritis (RA) is caused by immature osteoblasts disposed on the endosteal surface adjacent to the seat of the inflammation and bearing RANKL. The greater immaturity of these cells is associated with increased expression of RANKL and thus with higher capacity of stimulating the osteoclast lineage, bearing the RANK receptor (Fig. 3). The maturation of osteoblasts adjacent to an inflammatory site slows by giving precedence to the typical markers of “early” osteoblast line (pre-osteoblasts) such as Runx2 and loco-regional activation of osteoclasts with subsequent erosive damage. The balance between RANKL and osteoprotegerin in arthritic microenvironment, capable to stimulate or slow down the osteoblastic ripening, determines the extent of erosive evolution. In patients suffering from RA, RANKL mRNA levels are higher compared with non-arthritic patients, while osteoprotegerin is reduced. In particular, this imbalance (RANKL increased/osteoprotegerin reduced) is evident at the pannus–bone interface [19]. The headquarters of the RANKL-osteoprotegerin “imbalance” is therefore the interface and not the synovial environment, as evidenced by the absence of differences between the two environments in RA and AS [20]. In RA, other cells, but osteoblasts, express RANKL, in particular activated T cells and fibroblasts of the synovial membrane [21]. In the case of inflammatory stage, osteoblasts mature losing the ability to activate the osteoclast and the result will be a greater reparative capacity through bone formation. We can therefore hypothesize that low inflammatory and erosive activity of the microenvironment may in part be dependent on the maximum maturation of osteoblasts. The activation of the RANK/RANKL system, in turn dependent on a number of environmental stimuli capable of acting on the osteoblast, including TNF-α, will induce the maturation phase of osteoclasts. The completion of osteoclast differentiation requires the recruitment of TNF receptor-associated factor (TRAF) 2, 5, and 6 and downstream activation of NF-kB, AP1 (c-fos) and NFATc1, the latter being the final key for the expression of critical genes such as those encoding β-integrin and calcitonin receptor [22–25]. The maturation of the osteoblast is accompanied by the expression of specific cellular factors: from RUNX2 (osteoblast progenitor) to collagen type 1 and ALP (intermediate osteoblast) to osteocalcin and bone sialoprotein (mature osteoblast that mineralizes the matrix).

Osteoclast–osteoblast relationship. a The activity of osteoclasts (with RANK receptor) is inversely proportional to the degree of maturation of osteoblasts (with RANKL receptor). b The system that produces neobone apposition is inhibited by Dkk1. c The TNF alpha/Dkk1/Wnt system in rheumatoid arthritis and enthesoarthritis. TNFα activates osteoclasts and Dkk1 but inhibits osteoblasts. In rheumatoid arthritis, Dkk1 is very present in the inflammatory sites with consequent inhibition of Wnt signaling. On the contrary, in enthesoarthritis, Dkk1 has a low presence, and therefore the local Wnt system is active. d The overall effect of PGE2 favors neobone formation, and nonsteroidal anti-inflammatory drugs (NSAD) may adversely affect this effect
The role of TNF-α and other cytokines
TNF-α, released loco-regionally by tissue macrophages, synoviocytes, and activated T cells, is able to recruit directly pre-osteoclasts up-regulating the expression of RANK on them and inhibit the osteoblastic line, promoting the expression of Runx2 (pre-osteoblastic activity) and RANKL on their surface membrane and inhibiting that of alkaline phosphatase, osteocalcin, and collagen type I (mature osteoblastic activity). The overall effect is that of a greater, and consequently erosive, osteoclast activity.
The well-known clinical efficacy of anti-TNF-α agents in the treatment of AS confirms a clear pathogenic role of this cytokine in this disease. Importantly, these medications are effective in stopping the progression of radiological disease (both erosive and neobone appositive), especially in the first 2 years of treatment. But in subsequent years, the process of bone neoformation that leads to the synthesis of syndesmophytes seems to be less influenced [26–28]. This finding is not easily explained. Also, it is not yet demonstrated whether the anti-TNF-α agents started in very early stages of disease are capable to inhibit or slow down in a more or less effective way the neoformation of syndesmophytes [29, 30]. On the contrary, it should be considered that celecoxib seems to have a protective role on the development over time of syndesmophytes when used in a continuous manner [31–33]. What assumption can we make? A fact from which we could start is that the inflammation in the course of AS if not pharmacologically treated decreases and causes bone formation (syndesmophytes). Another fact to consider is that in lesser intense or more long-lasting inflammation, the neoappositive phase will start earlier. Therefore, it seems that the syndesmophytes form after inflammation is reduced or turned off and that the inflammatory process is necessary when turned off to ensure that neobone formation arises, but it seems that the two processes are independent. TNF-α plays a key role in the induction of the inflammatory erosive phase in the course of AS through some mechanisms. Direct osteoclast activation and osteoblast inhibition through DKK1 is a specific target of TNF-α in particular at the gene level and is evidenced in the sites of RA more than in those of AS [34]. It can be hypothesized that TNF-α is elevated in RA and in AS. In RA, TNF-α stimulates high DKK1 expression causing an overall frankly erosive effect (Wnt system locked). In contrast, the low DKK1 expression found in AS would lead to the absence of modulation by Wnt (Wnt active system) with two consequences: from one side an erosive process altogether less aggressive and from the other side the maintenance of a process of bone neoformation independent from TNF-α mediated activities (Fig. 3b, c). Therefore, the use of anti-TNF-α agents in a microenvironment such as that of AS, where DKK1 is low, would not produce a significant effect on the Wnt system compared with RA. WNT signaling remains hyperactive promoting a constant bone formation that shows up with the formation of syndesmophytes despite therapy with these drugs. This mechanism of Wnt-mediated bone formation is not the only being active in AS. Two additional systems may contribute to bone formation in AS: TGF-β and some members of its superfamily as BMPs. The latter, activated in chondrocytes by TNF-α and IL-1β, are over-expressed in DBA1 male mice that develop ankylosis, and their block by using specific inhibitors (nogging) prevents ankylosis. Importantly, BMPs are able to activate the Wnt signaling system [11, 35–40].
In a general vision, the anti-TNF-α agents have an osteoclast-blocking effect (with consequent blockage of erosive activity) and osteoblast-activating effect [41]. This would explain why, as already pointed out, despite the continuation of therapy with anti-TNF-α agents, the process of bone neoformation may tend to form syndesmophytes anyway, although an increase in bone reparative activity mediated by the blockade of TNF-α has not been demonstrated in humans [42, 43].
On the contrary, if the process of bone neoapposition was a direct consequence of the inflammatory erosive process, the block of the inflammatory phase would automatically block the neoappositive phase. IL-1 is secreted by macrophages and synovial fibroblasts of the microenvironment of RA and acts, in the cytokine network, downstream of the TNF-α mediating many functions. The increase in RANKL and osteoclast differentiation by the action of TNF-α are the effects mediated in part by IL-1 and its receptor (IL-1R). Moreover, the confirmation of the pro-erosive role of IL-1 comes from in vitro experiments. The breeding of arthritogenic hTNFTg mice and mice deprived of IL1 synthesis produces a progeny of mice with reduced osteoclastogenesis and less erosive aggressiveness [44–46]. However, IL1 block would result in less erosive aggressiveness, while maintaining the TNF-α-modulated arthritogenic microenvironment.
Other cytokines seem to play a key role in the inflammatory microenvironment of RA shifting the balance in favor of an up-regulation of RANKL and therefore of erosivity rather than bone neoformation. IL-6, synthesized by synovial macrophages and fibroblasts, on the one hand increases the expression of RANKL and therefore osteoclastogenesis and on the other hand activates Th17 cells, mediators, in turn, of the inflammatory process [47–49]. IL-17 also determines RANKL up-regulation and, at the same time, induces IL-6 expression [50].
A possible further adjustment system of the erosion/bone formation balance is represented by sclerostin triggered by mechanical inputs and mediated by the activation of osteocytes. The activation of this circuit would promote an overall pro-erosive action through inhibition of the Wnt pathway [51].
Another local system of regulation of erosion/bone formation balance is that of PGE2, mediator of an overall bone neoformative action biologically interpretable as “protective or reparative” of bone damage. This mechanism, active in human biology, is mediated, on the one hand, by a down-regulation of DKK1 and sclerostin, and secondly, by up-regulation of the Wnt pathway (Fig. 3d). As noted above, some studies have highlighted the possible role of celecoxib as long-term inhibitor of the formation of syndesmophytes taking advantage of inhibition of PGE2 circuit [31–33, 52].
The role of Wnt signaling
In erosive sites of RA, there is a bone neoapposition potential mediated by osteoblasts activated by Wnt signaling that is inhibited, however, by the increase in DKK1 in these sites. The inhibitors of DKK1 mitigate erosion by activating osteoprotegerin (OPG) [53]. The glycoproteins of Wnt family are derived from the transcription of Wg (Wingless) and INT genes. They act on specific cell membrane receptors activating intracellular pathways that in turn are able to activate cell differentiation in the osteoblastogenic sense [54]. Wnt signaling pathway is biologically oriented to bone neoformation. The Wnts 1 and 3 are synthesized by mesenchymal stem cells and pre-osteoblasts and depend on the presence of BMP2 as activating stimulus. Wnt signaling consists of two routes: the canonical and non-canonical [55]. When Wnts are free to act in the canonical way, they bind to specific membrane receptors: the Frizzled proteins and low-density lipoprotein receptor protein (LRP) 5 and 6. The next step is the phosphorylation of intracellular proteins Dsh and Axin by the membrane receptor system (LRP and Frizzled). The activation of Dsh and Axin allows the binding of the latter with the hlycogen synthase kinase (GSK-3)-β, which is inhibited and prevented from binding and inhibiting β-catenin, which is therefore free to move within the nucleus activating the transcription of Wnt target genes (Fig. 4a). When there is no signal of Wnts, β-catenin is “caged” by the Axin/Dsh/GSK-3 β protein complex resulting in phosphorylation of β-catenin with subsequent destabilization and degradation and prevention of gene transcription (Fig. 4b). Inhibitors of this canonical pathway are sclerostin and DKK1 that act on LRP 5 and 6, and at the membrane level, secreted frizzled-related protein 1 (SFRP1) which seizes the Wnts. The “activated” Canonical system of Wnt involves a metabolic system oriented to bone neoapposition. Conversely, if “inhibited” (for example with the presence in inflammatory sites of DKK1), the orientation of the microenvironment changes to erosive sense. The phenotype of osteoblasts deprived of β-catenin is comparable to that observed at the sites of bone erosion in the case of arthritis. This suggests that inhibition of canonical Wnt signaling pathway might be one of the mechanisms through which the neobone formation is impaired in the erosive seats of RA. l.

Wnt system of pre-osteoblasts and inhibition of the Wnt system. a When Wnts are free to act in the canonical way, they bind to specific membrane receptors, the Frizzled proteins and low-density lipoprotein receptor protein (LRP) 5 and 6. The next step is the phosphorylation of intracellular proteins Dsh and Axin by the membrane receptor system (LRP and Frizzled). The activation of Dsh and Axin allows the binding of the latter with the glycogen synthase kinase (GSK-3)-β, which is inhibited and prevented from binding and inhibiting β-catenin, which is therefore free to move within the nucleus activating the transcription of Wnt target genes. b When there is no signal of Wnts, β-catenin is “caged” by the Axin/Dsh/GSK-3 β protein complex resulting in phosphorylation of β-catenin with subsequent destabilization and degradation and prevention of gene transcription. Dkk1 and sclerostin block the LRP receptor
The non-canonical WNT signaling pathway, activated by Wnts 5, is parallel to the canonical and is based in fibroblasts of the synovial membrane of RA. It is not known whether it is also present in osteoblasts. The co-receptor membrane complex is constituted by the Frizzled proteins (receptor) and ROR2 and RYK (co-receptors). The activation of these membrane receptors comprises two parallel activations at the intracellular level: Wnt/Ca2+ pathway and planar cell polarity pathway (PCP). In the first case will be activated Nemo-like kinase (NLK) and Nuclear factor of activated T cells (NFAT), while in the second case the Rho family of GTPases (ROK) and c-Jun NH2 terminal kinase (JNK) with overall pro-transcriptive effect in the nucleus. The activated non-canonical pathway seems to be involved in erosions of RA by modulating, in the synovial membrane of this pathology, fibroblast activation, and the expression of RANKL. It is not known whether it is also active in osteoblasts of RA.
The FRP 1, 2, and 4 binding to Wnts block both these pathways by inhibiting the osteoblastic differentiation in certain diseases such as myeloma or steroid-related osteoporosis. DKK1 and 3 (Dickkopf proteins) block only the canonical way binding to LRP 5 and 6. Both inhibitors are up-regulated in tissues and erosive sites of RA in experimental animals. Only DKK1 is elevated in patients with RA and arthritic type hTNFTg mice [53]. After stimulation with TNF-α, DKK1 increases in synovial fibroblasts from arthritis in humans and arthritic mice. The block of DKK1 with specific antibodies reduces the erosive pattern of the joints in arthritic mice, increases the formation of juxta-articular osteophtes and is also associated with an up-regulation of OPG [14].
While FRP 1 and 4 seem to be expressed more in the fibroblasts, FRP 3 is represented in macrophages even if it is not clear the motivation of this different cellular distribution [56].
It must be emphasized that oxidative stress and hypoxic conditions, both common factors of the microenvironment of RA, are able to influence the erosive evolution inhibiting Wnt signaling, in the first case sequestering β-Catenin and in the second increasing DKK1 [57–59].
Role of Wnt system in AS
The environment of inflamed diarthrodial joints in seronegative spondyloarthritis is similar to that of RA with sub-chondral erosions, hyperplasia of the synovium, lymphomonocytic infiltration (mainly) of synovial tissues and neovascularization. The location and the extent of inflammation are usually elements that characterize the clinical diversity of the two diseases. The hallmark of AS is enthesitis with subsequent formation of syndesmophytes in the spine. The role of the Wnt signaling pathway is under judgment. It is possible that neobone formation characterizing enthesitic sites depends on Wnt pathway activation. In fact, the block of DKK1 in models of arthritic hTNFTg mice brings both to the formation of osteophytes where there were erosive sites [14] and ankylosis of the sacroiliac joint [60]. An animal model for the study of AS, the DBA1 mouse, shows arthritis associated with spontaneous joint fusion. BMPs (that activate the Wnt signaling pathway) seem to play a critical role in this regard [10]. In the same model of AS in mouse, the use of inhibitors of TNF-α does not affect the evolution of bone formation by excluding, at least in this model, a pathogenic role of TNF-α [12]. The Wnt system could therefore play an important role in the imbalance between bone formation (which prevails in the microenvironment of AS) and osteoresorption (which prevails in RA) [14, 60]. The mechanism that, in vivo, would release the Wnts might be, as pointed out, the very low concentration of DKK1 in inflammatory sites of AS.
The role of RANKL-RANK system
In the arthritic microenvironment, the imbalance between RANKL and osteoprotegerin (OPG), in favor of the former, determines evolution and magnitude of erosions. In RA, RANKL is expressed on the surface of osteoblasts and by activated T cells and fibroblasts of the synovial membrane [21].
In patients with RA, RANKL mRNA levels are higher compared with non-arthritic subjects or subjects with non-inflammatory phase, while OPG is more reduced. Therefore, the activation of osteoclasts is favored in this microenvironment. This imbalance is particularly evident in the “pannus—bone” interface [19] which seems to be the place where the “battle” in the course of inflammatory arthritis is recorded. In contrast, the articular environments of RA and AS do not express significant differences in RANKL: OPG equilibrium [20].
The signal required for the development of “erosive phase” both in RA than SpA is RANKL-RANK binding. The proof is that by blocking this signal with RANK.Fc in “artritogenic” hTNF-Tg mouse immature osteoclasts (CD11b+) accumulate, mature ones minimize on the spot and erosion blocks. This takes place despite a constitutional increase of TNF-α signaling which contributes to increase the number of osteoclast precursors [61].
Other locking mechanisms of RANKL theoretically with anti-erosive effectiveness are represented by osteoprotegerin–immunoglobulin Fc segment complex (OPG-Fc), with reduction in the number of osteoclasts at the pannus–bone interface [62], and denosumab, anti-RANKL humanized IgG2 monoclonal antibody [63].
The role of the TNAP system
TNF-α and IL-1β stimulate the activity of tissue non-specific alkaline phosphatase (TNAP) and mineralization in cultures of smooth muscle cells. In vivo these cytokines appear to be responsible for vascular calcification of atherosclerosis and type 2 diabetes mellitus. Instead, these cytokines inhibit TNAP activity in enthesitic chondrocytes in situ and in human and mouse chondrocytes in vitro. This different behavior seems to be due to a different mediation, in relation to the tissues, by the peroxisome proliferator-activated receptor (PPAR)-γ system. Therefore, in AS, the activation of this system mediated by the inflammatory process would inhibit enthesitic mineralization. TNF-α and IL-1β are therefore powerful and direct inhibitors of the formation of syndesmophyte in AS. Also for this reason, the anti-TNFα agents, while improving the symptoms of patients with AS, would not be able to control the neobone formation of syndesmophytes [64].
The role of the environment and genetics
Environmental factors certainly play a role in the pathogenesis of AS. However, if we consider that first-degree relatives of people with a AS have a risk of developing SpA 40 times higher than that of the general population, and that the possibility of developing AS in twins HLA-B27+ is 23 % in dizygotic twins and 63 % in monozygotic twins, we recognize the important role of inheritance [65].
The association with HLA-B27 was theorized in 1970, and its pathogenic role is not at all in doubt. 90 % of AS expressing HLA-B27 and 5 % of the population will develop an HLA-B27+ AS. These data lead to the hypothesis that some variables related to HLA-B27 or, on the contrary, other genes or gene polymorphisms outside of major histocompatibility complex (MHC) may play a pathogenic role. In the international literature, four genes come out as candidates to play a role in the pathogenesis of AS: HLA-B27, the cluster of IL-1, the ARTS1 gene, and the IL-23R gene.
Hla-b27
HLA-B27 gene is related to a risk of developing AS of 20–50 % [66]. This risk is reduced in non-caucasian populations (Lebanon, black populations in West Africa and Indonesia) and in patients with concurrent psoriasis and inflammatory bowel diseases. We recognize 45 variants of HLA-B27 on the basis of the nucleotide sequence. The most common variant in the Caucasian population is the ancestral HLA-B*2705, followed by HLA-B*2702. Other variants prevail in other populations. The Sardinian population seems to dominate the HLA-B*2709 although probably it has no causative role in AS [67]. The issue of the HLA-B*2709 is, however, a bit more complex. Some individuals with this allele are affected by spondyloarthritis but not from AS [68], and two patients with AS were described [69, 70]. Besides, in Sardinia, the HLA-B*2705 and HLA-B*2709 variants segregate into two different haplotypes, making possible the hypothesis that HLA not B27+ have a modulatory role on the loss of the associative relationship between AS and the HLA-B*2709 variant [71]. Furthermore, some genes related to the killer function seem to have a protective role on the development of AS in HLA-B*2709+ subjects [72]. Therefore, the role of HLA-B*2709 variant is currently not fully understood. The HLA-B*2706 variant, despite prevailing in the populations of southeast Asia, does not seem to be associated with the development of AS [73]. Both HLA-B*2709 and HLA-B*2706 variants differ from HLA-B*2705 variant for an amino acid change at position 116, area with a high variability of the heavy chain. This small diversity strongly alters the binding capacity of the HLA molecule to the hypothetical arthritogenic peptide characterized, in turn, by the presence of a tyrosine residue in position P9. Such position and that in P2 may represent a critical site for the binding between the peptide and the HLA molecule (adhesion amino acids in the pocket of the HLA molecule) [67]. In addition to HLA-B*2705, HLA-B*2702 and also the HLA-B*2704 appear to be associated with AS in many populations [74]. The HLA-B*2707, present in many populations, seems to have a modulation role more than being a true genetic risk factor for developing AS [75]. Ultimately, because only 5 % of the population will develop a HLA-B27+ SpA? One reason is certainly the non-pathogenicity of some HLA-B27 variants (HLA-B*2709 and HLA-B*2706). And for variants notoriously pathogenic such as the HLA-B*2705? In this case one could hypothesize the need to have a high degree of molecular expression of HLA class I molecules on the surface of the antigen-presenting cell (APC) in order to have an effective modulating function on antigen presentation [76–81].
At the moment, the hypotheses about the role of HLA-B27 in the pathogenesis of AS are five:
-
a.
Theory of the arthritogenic peptide. HLA class I antigens (such as HLA-B27) positioned on the APCs have the role of presenting antigens to CD8+ T cells. In particular, crystallographic studies carried out on the HLA-B*2705 have demonstrated the presence of “pockets” (from A to F) binding the antigenic peptide with specific locations able to bind the complementary sequence of the amino- or carboxy terminus of the bound peptide. The “B” pocket is the same for all variants of HLA-B27 but changes in the HLA-B antigens, where there is an amino acid (glutamine) at position 45 that binds a second amino acid (arginine) at position 2 conferring specificity to the pocket relative to the type of peptide to bind. The variant HLA-B*2709 and HLA-B*2706, as already pointed out, when compared to HLA-B*2705 have only one different amino acid in the pocket “F” that binds the C-terminal portion of the amino acidic peptide. These differences between the variants will theoretically cause a different ability to bind with antigenic peptides. This must be the assumption on which to base our discussion. This immune response, restricted against an environmental peptide, would probably occur against microbial antigens like what happens in reactive arthritis, toward which the AS has many similarities.
-
b.
Theory of “molecular mimicry.” In this case, a self-reactive immunity mediated by cytotoxic T lymphocytes would develop against self-antigens that express antigenic homology along with microbial agents. Klebsiella pneumoniae and Shigella may be some of the accountable microorganisms having sequence homology with some HLA-B27 variants [82, 83]. HLA-derived peptides may also be presented as antigens by the molecules of class I and II [84].
-
c.
Theory of “misfolding.” The heavy chains of HLA-B27 are often assembled in a problematic way in the reticuloendothelial system resulting in their accumulation with secondary “stress” and consequent activation of NF-κB which, in turn, leads to the synthesis of pro-inflammatory cytokines (TNF α) by the macrophage/monocyte system. This theory entails a pathogenic role for HLA-B27 regardless of hypothetical environmental arthritogenic peptides [85].
-
d.
Thymic selection. The HLA-B27 may select, at the thymic level, restricted CD8+ T lymphocyte clones specific for microbial antigens [86].
-
e.
Theory of heavy chains. The heavy chains of HLA-B27, not complexed with β-2-microglobulin, would be expressed on the cell surface behaving like HLA class II molecules. They would interact with CD4+ helper T lymphocytes [87, 88].
The cluster of IL-1
The cluster of IL-1 comprises nine genes on chromosome 2, including the gene encoding IL-1α, IL-1β, and IL1 receptor antagonist (IL1-RN). The possible correlation between AS and the polymorphism of IL1-RN allele has not been confirmed in recent studies, whereas there is confirmation for IL-1α and IL-1β [89–91]. The relative risk of developing AS in association with genetic variants related to these cytokines is of the order of 4–6 %. IL-1α is produced by macrophages and has a pro-inflammatory action, but its role in the pathogenesis of AS is not known [92], although the clinical efficacy in AS of its inhibitor, anakinra, although lower respect to anti-TNFα agents, suggests its involvement [93, 94].
The ARTS1 gene
The risk of developing AS in relation to this gene is 26 %. ARTS1 encodes an amino peptidase of the endoplasmic reticulum which has two main functions. The first is the cleavage of the receptors of IL1, IL6, and TNFα on the cell surface, so that the loss of its function has pro-inflammatory effects. The second is the cleavage of the N-terminal portion of the precursor of the arthritogenic peptide at the level of the reticulum so as to make it appropriate to the presentation of HLA class I molecules. A defect of such a function would lead to a modulation of the presentation of the peptide to the APC. This assumption, although not proven, would affect the existence of specific arthritogenic peptides confined to the intracellular processing [95, 96].
The IL23R gene
The IL23R gene seems to be correlated to psoriasis and inflammatory bowel involvement, but at the same time, there is evidence of correlation with AS [97, 98]. IL23 belongs to the IL12 family, and one of the two constituent subunits (p9 and p40), the p40, is an integral part of IL12. The IL23 is produced by the activation of APCs and has the role of keeping the orientation of immunological CD4+ naive T cells toward Th17 cells, independent from the Th1 and Th2 cells, which express the IL23R thanks to IL6 and TGF-β and which release IL-17, a pro-inflammatory cytokine able to induce the synthesis of IL1, IL6, and TNF-α [97, 98]. The activation of the IL23/IL17 axis is present both in the type of innate immune mechanisms (with activation of receptors such as the Toll-like receptor 4) and in the adaptive type (for example in response to infection) and seems to have a role, not yet fully clarified, in the pathogenesis of AS [99, 100]. The effectiveness of anti-IL23 (ustekinumab) [101–103] would confirm a pathogenic role of this cytokine in both AS in psoriasis and Crohn’s disease [98]. A fact to note is the low concentration of INF-γ in monocytes and in circulating T cells in the course of AS compared with other clinical inflammatory conditions [104, 105]. The low value of INF-γ could theoretically affect an easy persistence of infection, and on one side, this could trigger the activation of the pathogenic mechanisms of AS and on the other hand could represent a stimulus for the activation of Th17 cells with release of IL-17 [106].
Summary of the physiopathogenetic model
The structural damage of AS is characterized by excessive neobone formation, with the syndesmophyte as a typical lesion. The process behind their formation is not yet fully understood although there are many useful information to clarify the physiopathogenetic puzzle.
The primum movens of the enthesitic process would be the micro-trauma to which entheses are subject, especially in the lower limbs, for biomechanical reasons. The inflammatory process would be facilitated by the sequential structure of the organ enthesis, constitutionally devoid of sub-enthesitic cortical bone and therefore closely related to the underlying trabecular bone and the medullary vascular system. The reparative attempt from the vascular system, thanks to the activating action of certain loco-regional cytokines, such as TNF α, would condition the possible deposit in the entheses of bacterial molecules derived from other organic sites and be able, especially in HLA-B27+ subjects, to activate an immune-mediated inflammatory process following the initial mechanical process. In this way, the inflammatory process, initially forced to fade gradually, could self-renew.
Imaging studies point out that the formation of syndesmophyte requires a local inflammatory process. It also seems that this neobone appositive process can be activated especially when the enthesitic inflammation is turned off. Furthermore, the formation of syndesmophytes in sites not previously involved by a radiologically detectable inflammatory process could theoretically be explained by the presence of a sub-expressed inflammation, more evident at the histochemical than at the radiological level [107]. But if the two processes, inflammatory and neobone appositive, are interdependent because the second is the necessary consequence of the first, the starting mechanism must be unique. The role of inflammatory cytokines, and TNF-α in particular, is precisely to start the process in its entirety. Depending on whether the process is activated in a pathological/clinical set rather than in another, the response is not always superimposable.
In fact, if in the joints and under physiological conditions there is a substantial balance between anabolic and catabolic processes of bone and cartilage, in rheumatic diseases, such as rheumatoid arthritis and AS, there is a “decoupling” between bone formation and bone resorption with final effect of loss (=erosion) in rheumatoid arthritis or neoapposition (syndesmophyte) in AS. But who makes this de-coupling? The answer is cytokines and factors released in the microenvironment of the joint. The pro-inflammatory cytokines regulate the status of inflammation in rheumatoid arthritis and AS, and also the expression of the factors that modulate the local bone remodeling. It is obvious that, being similar the cytokines in both processes, the different evolutionary outcomes should depend on the articular inflammatory microenvironment. At the moment, the main effectors of the microenvironment responsible of these differences are related to the balance in the expression of RANKL: OPG (critical for modulating osteoclast activity) and the balance of activation or inhibition of Wnt signaling (critical for modulating osteoblast activity but with potential effects on both bone formation and resorption).
Both of these systems are in fact modulated by TNF-α, with direct effects of activators (osteoclasts, DKK1, BMPs, and TGF-β) or inhibitors (osteoblasts, TNAP) and indirect inhibitory effects (Wnt)
There are two possible explanations of the reasons for the different evolution of the inflammatory microenvironment of rheumatoid arthritis (dominated by a process of erosion and suppression of the reparative) compared to that of AS (in which the erosion process is minor and accompanied by a gradual emergence of the healing process).
Firstly, if TNF-α is abundantly expressed on site in both diseases, DKK1 (one of the main targets of TNF-α) is much less expressed in the microenvironment of AS, implying a substantial irrelevance of TNF-α on Wnt system in this pathology compared to rheumatoid arthritis. The Wnt system would therefore not be totally inhibited (activating stimulus of BMPs in turn activated by TNF-α, decreased inhibitory stimulus of DKK1 depressed and activating stimulus of PGE2), and this would affect the lower erosive profile that the inflammatory phase shows in AS compared with rheumatoid arthritis.
Secondly, it was hypothesized that if the inflammation of rheumatoid arthritis is persistent with complete suppression of the repair process for the dominant activity of RANKL, DKK1, and sclerostin system, in AS inflammation may be fluctuating. In this case, there may be temporal spaces for reparative phenomena mediated by the system of PGE2, Wnt, BMP2, and TNAP already active, albeit in a latent manner, in the inflammatory phase [108, 109]. The presence at nuclear magnetic resonance imaging of focal lesions of the fat tissue in the vertebral corners, sites of the enthesitic process, could be a useful element to identify early activation of an anabolic phase in the enthesitic process as expression of an early attempt to repair [110, 111].
In this scenario of fluctuating inflammation, early initiation of therapy with anti-TNF-α agents could avoid by anticipating the onset, the anabolic phase during the switch between early active phase and a resting inflammation. In this sense, it could be valuable to start the therapy with anti-TNF-α agents very early. This choice, coupled with a longer observational follow-up, showed a greater ability of anti-TNF-α agents to limit the formation of syndesmophyte, without completely inhibiting the synthesis [29, 30].
The simultaneous presence in the enthesitic sites of osteogenic and condrogenic areas evidenced by histological studies may express the incomplete capacity of TNF-α to avoid bone neoapposition with reparative function already during the inflammatory phases of the disease. This condition may result from the active expression of BMP2, the presence of which is stimulated by TNF-α and by lack of inhibition of Wnt ostogenic boost by DKK1. Once the inflammatory phase resolves spontaneously or with anti-TNF-α agents, the braking action of TNF-α on Wnt system and TNAP system would weaken, and together with the intrinsic reparative conditions put in place by the organ enthesis tissue, would accelerate the reparative enthesopatic function (TNF-α brake hypotesis) [112].
Conclusions
The pathological conditions defined as seronegative spondyloarthritis or enthesoarthritis are featured by distinctive osteoarticular and extra-articular manifestations, but different diseases included in this group can affect different members of the same family and may show diverse aspects of the various phenotypic disease in the same patient, with clinical overlaps hindering the differential diagnosis. The inflammatory process in the synovio-entheseal sites is defined as enthesitis that represents an anatomo-pathological hallmark of enthesoarthritis and is characterized by an initial inflammatory/erosive phase, followed by a phase of neobone apposition. This phase appears to advance autonomously from the preceding erosive phase implying that different hinges trigger the physiopathogenetic mechanisms underlying the two processes. The poorer control exerted by anti-TNF-α agents on the enthesopatic neobone appositive phase respect to the inflammatory/erosive phase could be explained by elucidate the different mechanisms driving these pathological processes.
References
Benjamin M, McGonagle D. The anatomical basis for disease localisation in seronegative spondyloarthropathy at entheses and related sites. J Anat. 2011;199:503–26.
Benjamin M. The enthesis organ concept and its relevance to the spondyloarthropathies. In: Lopez-Larrea C, Diaz-Pena R, editors. Molecular mechanisms of spondyloarthropathies, vol. 4. New York: Landes Bioscience and Springer Sciences Business Media; 2009. p. 57–70.
Benjamin M, Toumi H, Suzuki D, Redman S, Emery P, McGonagle D. Microdamage and altered vascularity at the enthesis-bone interface provides an anatomic explanation for Bone involvement in the HLA-B-27-associated spondyloarthritides and allied conditions. Arth Rheum. 2007;56(1):224–33.
McGonagle D, Lories RJ, Tan AL, Benjamin M. The concept of a “synovio-entheseal complex” and its implications for understanding joint inflammation and damage in psoriatic arthritis and beyond. Arth Rheum. 2007;56:2482–91.
Kahn MF, Chamot AM. SAPHO syndrome. Rheum Dis Clin North Am. 1992;18:225–46.
Chamot AM, Benhamou CL, Kahn MF, Beraneck L, Kaplan G, Prost A. Acne-pustolosis-hyperostosis-osteitis syndrome: results of a National survey, 85 cases (review). Rev Rhum Mal Osteoartic. 1987;54:187–96.
McGonagle D, Stockwin L, Isaacs J, Emery P. An enthesitis based model for the pathogenesis of spondyloathropathy: additive effects of microbial adjuvant and biomechanical factors at disease sites. J Rheumatol. 2001;28:2155–9.
Maksymowych WP. Ankylosing spondylitis: at the interface of bone and cartilage. J Rheumatol. 2000;27:2295–301.
McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. 2006;3(8):e297.
Lories RJ, Derese I, Luyten FP. Modulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing enthesitis. J Clin Invest. 2005;115:1571–9.
Lories RJ, Daans M, Derese I, et al. Nogging haploinsufficiency differentially affects tissue responses in destructive and remodeling arthritis. Arth Rheum. 2006;54:1736–46.
Lories RJ, Derese I, de Bari C, Luyten FP. Evidence for uncoupling of inflammation and joint remodelling in a mouse model of spondylarthritis. Arth Rheum. 2007;56:489–97.
Lories RJ, Derese I, Luyten FP. Inhibition of osteoclast does not prevent joint ankylosis in a mouse model of spondyloartrhritis. Rheumatology. 2008;47:605–8.
Diarra D, Stolina M, Polzer K, et al. Dickkopf-1 is a master regulator of joint remodelling. Nat Med. 2007;13:156–63.
Uderhardt S, Diarra D, Katzenbeisser J, et al. Blockade of Dickkopf (DKK)-1 induces fusion of sacroiliac joints. Ann Rheum Dis. 2010;69(3):592–7. doi:10.1136/ard.2008.102046.
Keffer J, Probert L, Cazlaris H, et al. Transgenic mice expressing human tumor necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–31.
Shealy DJ, Wooley PH, Emmell E, et al. Anti-TNF alpha antibody allows healing of joint damage in polyarthritic transgenic mice. Arthritis Res. 2002;4:R7.
Pettit AR, Ji H, von Stechow D, et al. TRANCE/RANKL knockout mice are protected from bone erosionin a serum transfer model of arthritis. Am J Pathol. 2001;159:1689–99.
Haynes DR, Barg E, Crotti TN, et al. Osteoprotegerin expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathies and osteoarthritis and normal controls. Rheumatology. 2003;42:123–34.
Vandooren B, Cantaert T, Noordenbos T, Tak PP, Baeten D. The abundant synovial expression of the RANK/RANKL/Osteoprotegerin system in peripheral spondylarthritis is partially disconnected from inflammation. Arthritis Rheum. 2008;58:718–29.
Kotake S, Udagawa N, Hakoda M, et al. Activated human T cells directly induce osteoclastogenesis from human monocytis: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum. 2001;44:1003–12.
Crotti TN, Flannery M, Walsh NC, Fleming JD, Goldring SR, McHugh KP. NFATc1 directly induces the human beta3 integrin gene in osteoclast differentiation. J Muscoloskeletal Neuronal Interact. 2005;5:335–7.
Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473:139–46.
Crotti TN, Sharma SM, Fleming JD, et al. PU.1 and NFATc1 mediate osteoclastic induction of the mouse beta3 integrin promoter. J Cell Physiol. 2008;215:636–44.
Shen Z, Crotti TN, Flannery MR, Matsuzaki K, Goldring SR, McHugh KP. A novel promoter regulates calcitonin receptor gene expression in human osteoclasts. Biochim Biophys Acta. 2007;1769:659–67.
van der Heijde D, Landewé R, Einstein S, et al. Radiographic progression of ankylosing spondylitis after up to two years of treatment with etanercept. Arthritis Rheum. 2008;58:1324–31.
van der Heijde D, Landewé R, Baraliakos X, et al. Radiographic findings following two years of infliximab therapy in patients with ankylosing spondylitis. Arthritis Rheum. 2008;58:3063–70.
Van der Heijde D. Adalimumab therapy for ankylosing spondylitis over two years does not demonstrate inhibition of radiographic progression compared with a historical control group. Arthr Rheum 2008;58(Suppl):5413; abstract 670.
Baraliakos X, Listing J, Brandt J, et al. Radiographic progression in patients with ankylosing spondylitis after 4 yrs of treatment with the anti-TNF-alpha antibody infliximab. Rheumatology (Oxford). 2007;46:1450–3.
Braun J, Baraliakos X, Listing J, et al. Persistent clinical efficacy and safety of anti-tumour necrosis factor alpha therapy with infliximab in patients with ankylosing spondylitis over 5 years: evidence for different types of response. Ann Rheum Dis. 2008;67(3):340–5.
Wanders A, Dv Heijde, Landewé R, et al. Non steroidal anti-inflammatory drugs induce radiographic progression in patients with ankylosing spondylitis: a randomized clinic trial. Arthritis Rheum. 2005;52:1756–65.
Boersma JW. Retardation of ossification on the lumbar vertebral column in ankylosing spondylitis by means of phenylbutazone. Scan J Rheum. 1976;5:60–4.
Krischak GD, Augat P, Blakytny R, Claes L, Kinzl L, Beck A. The non steroidal anti inflammatory drug diclofenac reduces appearance of osteoblasts in bone defect healing in rats. Arch Orthop Trauma Surg. 2007;127:453–8.
Qian J, Xie J, Hong S, et al. Dickkopf-1 (DKK1) is a widely expressed and potent tumor associated antigen in multiple myeloma. Blood 2007;110:1587–1594.
Claudepierre P, Wendling D. Are inflammation and ossification on separate tracks in ankylosing spondylitis? Joint Bone Spine. 2008;75:520–2.
François RJ, Neure L, Sieper J, Braun J. Immunohistological examination of open sacroiliac biopsies of patients with ankylosing spondylitis: detection of tumor necrosis factor alpha in two patients with early disease and transforming growth factor beta in three more advanced cases. Ann Rheum Dis. 2006;65:713–20.
Braun J, Bollow M, Neure L, et al. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arth Rheum. 1995;38:499–505.
Archer JR. Ankylosing spondylitis, IgA, and transforming growth factors. Ann Rheum Dis. 1995;54:544–6.
Claudepierre P, Rymer JC, Authier FJ, et al. A relationship between TGF-beta 1 or IL-6 plasma levels and clinical features of spondyloarthropathies. Br J Rheumatol. 1997;36:400–1.
Wendling D, Cedoz JP, Racadot E, Dumoulin G. Serum IL17, BMP-7, and bone turnover markers in patients with ankylosing spondylitis. Joint Bone Spine. 2007;74:304–5.
Roux S, Orcel P. Bone loss. Factors that regulate osteoclast differentiation: un up-date. Arthritis Res. 2000;2:451–6.
Li P, Schwarz EM, O’Keefe RJ, et al. Systemic tumor necrosis factor alpha mediates an increase in peripheral CD11b high osteoclast precursor in tumor necrosis factor alpha-transgenic mice. Arthritis Rheum. 2004;50:265–76.
Kaneki H, Guo R, Chen D, et al. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. J Biol Chem. 2006;281:4326–33.
Ji H, Pettit A, Ohmura K, et al. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induces arthritis. J Exp Med. 2002;196:77–85.
Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. 2005;115:282–90.
Zwerina J, Redlich K, Polzer K, et al. TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci USA. 2007;104:11742–7.
Fonseca JE, Santos MJ, Canhão H, Choy E. IL6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev. 2009;8:538–42.
Kato A, Matsuo S, Takai H, Uchiyama Y, Mihara M, Suzuki M. Early effects of tocilizumab on bone and bone marrow lesions in a collagen-induced arthritis monkey model. Exp Mol Pathol. 2008;84:262–70.
Jones G, Sebba A, Gu J, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69(1):88–96.
Ogura H, Murakami M, Okuyama Y, et al. IL17 promotes autoimmunity by triggering a positive feed-back loop via IL6 induction. Immunity. 2008;29:628–36.
Poole KE, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–4.
Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42:606–15.
Walsh NC, Reinwald S, Manning CA, et al. Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. J Bone Miner Res. 2009;24:1572–85.
Bodine PV, Komm BS. Wnt signaling and osteoblastogenesis. Rev Endocr Metab Disord. 2006;7:33–9.
Moon RT, Kohn AD, De Ferrari GV, Kaykas A. Wnt and beta-catenin signaling: diseases and therapies. Nat Rev Genet. 2004;5:691–701.
Ijiri K, Nagayoshi R, Matsushita N, et al. Differential expression patterns of secreted frizzled related protein genes in synovial cells from patients with arthritis. J Rheumatol. 2002;29:2266–70.
Arnett TR, Gibbons DC, Utting JC, et al. Hypoxia is a major regulator of osteoclast formation and bone resorption. J Cell Physiol. 2003;196:2–8.
Utting JC, Robins SP, Brandao-Burch A, Orriss IR, Behar J, Arnett TR. Hypoxia inhibits the growth, differentiation and bone forming capacity of rat osteoblasts. Exp Cell Res. 2006;312:1693–702.
Brandao-Burch A, Utting JC, Orriss IR, Arnett TR. Acidosis inhibits bone formation by osteoblasts in vitro by preventing mineralization. Calcif Tissue Int. 2005;77:167–74.
Uderhardt S, Diarra D, Katzenbeisser J, et al. Blockade of Dickkopf 1 induces fusion of sacroiliac joints. Ann Rheum Dis. 2010;69(3):592–7.
Li P, Schwarz EM, O’Keefe RJ, Ma L, Boyce BF, Xing L. RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclasts precursor but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J Bone Miner Res. 2004;19:207–13.
Redlich K, Hayer S, Maier A, et al. Tumor necrosis factor alpha-mediated joint destruction is inhibited by targeting osteoclast with osteoprotegerin. Arthritis Rheum. 2002;46:785–92.
Cohen SB, Dore RK, Lane NE, et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 2008;58:1299–309.
Lencel P, Delplace S, Pilet P, et al. Cell-specific effects of TNF-alpha and IL-1 beta on alkaline phosphatase: implication for syndesmophyte formation and vascular calcification. Lab Invest. 2001;91:1434–42.
Pham T. Pathophysiology of ankylosing spondylitis: what’s new? Joint Bone Spine. 2008;75:656–60.
Brown MA, Kennedy LG, MacGregor AJ, et al. Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum. 1997;40:1823–8.
D’Amato M, Fiorillo MT, Carcassi C, et al. Relevance of residue 116 of HLA-B27 in determining susceptibility to ankylosing spondylitis. Eur J Immunol. 1995;25:3199–201.
Ramos M, López de Castro JA. HLA-B27 and the pathogenesis of spondyloarthritis. Tissue Antigens. 2002;60:191–205.
Cauli A, Vacca A, Mameli A, et al. A Sardinian patient with ankylosing spondylitis and HLA-B*2709 co-occurring with HLA-B*1403. Arthritis Rheum. 2007;56:2807–9.
Olivieri I, D’Angelo S, Scarano E, Santospirito V, Padula A. The HLA-B*2709 subtype in a woman with early ankylosing spondylitis. Arthritis Rheum. 2007;56:2805–7.
Fiorillo MT, Cauli A, Carcassi C, et al. Two distinctive HLA haplotypes harbor the B27 alleles negatively or positively associated with ankylosing spondylitis in Sardinia: implications for disease pathogenesis. Arthritis Rheum. 2003;48:1385–9.
Cascino I, Paladini F, Belfiore F, et al. Identification of previously unrecognized predisposing factors for ankylosing spondylitis from analysis of HLA-B27 extended haplotypes in Sardinia. Arthritis Rheum. 2007;56:2640–51.
López-Larrea C, Sujirachato K, Mehra NK, et al. HLA-B27 subtypes in Asian patients with ankylosing spondylitis. Evidence for new associations. Tissue antigens. 1995;45:169–76.
Gonzalez-Roces S, Alvarez MV, Gonzalez S, et al. HLA-B27 polymorphism and worldwide susceptibility to ankylosing spondylitis. Tissue Antigens. 1997;49:116–23.
Varnavidou-Nicolaidou A, Karpasitou K, Georgiou D, et al. HLA-B27 in the Greek Cypriot population: distribution of subtypes in patients with ankylosing spondylitis and other HLA-B27-related diseases. The possible protective role of B*2707. Human Immunol. 2004;65:1451–4.
Sprent J, Schaefer M. Antigen presenting cells for CD8+ cells. Immunol Rev. 1990;117:213–34.
Taurog JD, Maika SD, Simmons WA, Breban M, Hammer RE. Susceptibility to inflammatory disease in HLA-B27 transgenic rat lines correlates with the level of B27 expression. J Immunol. 1993;150:4168–78.
Cauli A, Dessole G, Passiu G, Mathieu A. Quantification of cellular antigens by means of flow cytometry and its role in rheumatology. Reumatismo. 2001;53:14–7.
Poncelet P. Microbeads and flow cytometry: how and why put the “-metry” in immuno-cytometry? Ann Biol Clin (Paris). 2004;62(1):53–7.
Cauli A, Dessole G, Nurchis PP, et al. The role of HLA-B27 molecules in the pathogenesis of ankylosing spondylitis. Reumatismo. 2002;54(3):266–71.
Cauli A, Dessole G, Fiorillo MT, et al. Increased level of HLA-B27 expression in ankylosing spondylitis patients compared with healthy HLA-B27 positive subjects: a possible further susceptibility factor for the development of disease. Rheumatology (Oxford). 2002;41(12):1375–9.
Ebringer A, Ahmadi K, Fielder M, et al. Molecular mimicry: the geographical distribution of immune responses to Klebsiella in ankylosing spondylitis and its relevance to therapy. Clin Rheumatol. 1996;15:57–67.
Tsuchiya N, Husby G, Williams RC Jr, Stieglitz H, Lipsky PE, Inman RD. Autoantibodies to HLA-B27 sequence cross-react with the hypothetical peptide from the arthritis—associated Shigella plasmid. J Clin Invest. 1990;85:1193–203.
Fiorillo MT, Maragno M, Butler R, Dupuis ML, Sorrentino R. CD8+ T cell auto-reactivity to HLA-B27-restricted self-epitope correlates with ankylosing spondylitis. J Clin Invest. 2000;106:47–53.
Mear JP, Schreiber KL, Münz C, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol. 1999;163:6665–70.
Lipsky PE. Spondyloarthopathies. In: Klippel JH, Dieppe PA, editors. Rheumatology 2nd ed. London: Mosby, 1999: 6.12.1–12.
Khare SD, Hansen J, Luthra HS, David CS. HLA-B27 heavy chains contribute to spontaneous inflammatory disease in B27/human beta 2-microglobulin (beta2 m) double transgenic mice with disrupted mouse beta2m. J Clin Invest. 1996;98:2746–55.
Allen RL, O’Callaghan CA, McMichael AJ, Bowness P. Cutting edge: HLA-B27 can form a novel beta 2-microglobulin-free heavy chain homodimer structure. J immunol. 1999;162:5045–8.
Kim TH, Stone MA, Rahman P, et al. Interleukin 1 and nuclear factor-kappaB polymorphism in ankylosing spondylitis in Canada and Korea. J Rheumatol. 2005;32:1907–10.
Sims AM, Timms AE, Bruges-Armas J, et al. Prospective meta-analysis of interleukin 1 gene complex polymorphisms confirm associations with ankylosing spondylitis. Ann Rheum Dis. 2008;67:1305–9.
Maksymowych WP, Rahman P, Reeve JP, Gladman DD, Peddle L, Inman RD. Association of the IL1 gene cluster with susceptibility to ankylosing spondylitis: an analysis of three Canadian populations. Arthritis Rheum. 2006;54:974–85.
Niki Y, Yamada H, Kikuchi T, et al. Membrane associated IL-1 contributes to chronic synovitis and cartilage destruction in human IL-1 alpha transegenic mice. J Immunol. 2004;172:577–84.
Tan AL, Marzo-Ortega H, O’Connor P, Fraser A, Emery P, McGonagle D. Efficacy of anakinra in active ankylosing spondylitis: a clinical and magnetic resonance imaging study. Ann Rheum Dis. 2004;63:1041–5.
Haibel H, Rudwaleit M, Listing J, Sieper J. Open label trial of anakinra in active ankylosing spondylitis over 24 weeks. Ann Rheum Dis. 2005;64:296–8.
Yan J, Parekh VV, Mendez-Fernandez Y, et al. In vivo role of ER-associated peptidase activity in tailoring peptides for presentation by MHC class Ia and class Ib molecules. J Exp Med. 2006;203:647–59.
Hammer ND, Schmidt JC, Chapman MR. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc Natl Acad Sci USA. 2007;104:12494–9.
McGovern D, Powrie F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut. 2007;56:1333–6.
Wendling D. Interleukin 23: a key cytokine in chronic inflammatory disease. Joint Bone Spine. 2008;75:517–9.
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukin 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9.
Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–9.
Krueger GG, Langley RG, Leonardi C, et al. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med. 2007;356:580–92.
Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial. (PHOENIX 2). Lancet. 2008;371:1675–84.
Goldminz AM, Gottlieb AB. Ustekinumab for psoriasis and psoriatic arthritis. J Rheumatol Suppl. 2012;89:86–9. doi:10.3899/jrheum.120253.
Rudwaleit M, Siegert S, Yin Z, et al. Low T cell production of TNF alpha and INF gamma in ankylosing spondylitis: its relation to HLA-B27 and influence of the TNF-308 gene polymorphism. Ann Rheum Dis. 2001;60:36–42.
Smith JA, Barnes MD, Hong D, DeLay ML, Inman RD, Colbert RA. Gene expression analysis of macrophages derived from ankylosing spondylitis patients reveals interferon-gamma dysregulation. Arthritis Rheum. 2008;58(6):1640–9.
Layh-Schmitt G, Colbert RA. The interleukin-23/interleukin-17 axis in spondyloarthritis. Curr Opin Rheumatol. 2008;20:392–7.
van der Heijde D, Machado P, Braun J, et al. MRI inflammation at the vertebral unit only marginally predicts new syndesmophyte formation: a multilevel analysis in patients with ankylosing spondylitis. Ann Rheum Dis. 2012;71(3):369–73.
Schett G, Rudwaleit M. Can we stop progression of ankylosing spondylitis ? Best Pract Res Clin Rheumatol. 2010;24(3):363–71.
Sieper J, Appel H, Braun J, Rudwaleit M. Critical appraisal of assessment of structural damage in ankylosing spondylitis: implications for treatment outcomes. Arthritis Rheum. 2008;58(3):649–56.
Hermann KG, Baraliakos X, van der Heijde DM, et al. Descriptions of spinal MRI lesions and definition of a positive MRI of the spine in axial spondyloarthritis: a consensual approach by the ASAS/OMERACT MRI study group. Ann Rheum Dis. 2012;71(8):1278–88.
Chiowchanwisawakit P, Lambert RG, Conner-Spady B, Maksymowych WP. Focal fat lesions at vertebral corners on magnetic resonance imaging predict the development of new syndesmophytes in ankylosing spondylitis. Arthritis Rheum. 2011;63(8):2215–25.
Maksymowych WP, Chiowchanwisawakit P, Clare T, Pedersen SJ, Østergaard M, Lambert RG. Inflammatory lesions of the spine on magnetic resonance predict the development of new syndesmophytes in ankylosing spondylitis: evidence of a relationship between inflammation and new bone formation. Arthritis Rheum. 2009;60(1):93–102.
Acknowledgments
We apologize for not citing many important references that have contributed to this review due to space limitations. The authors are grateful for support to their research from the “5 × 1000” voluntary contribution. None of the authors received any funding related to the writing of this manuscript, and the funding bodies did not play any role in the writing of the manuscript or decision to submit the manuscript for publication.
Conflict of interest
The authors have no conflict of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
De Cata, A., Inglese, M., Rubino, R. et al. The synovio-entheseal complex in enthesoarthritis. Clin Exp Med 16, 109–124 (2016). https://doi.org/10.1007/s10238-015-0341-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-015-0341-x