
Abstract
SP110 is a promising anti-Mycobacterium tuberculosis (MTB) gene. To investigate the effects of SP110 and its associated genes, i.e., MYBBP1A and RELA, on pathological progression of MTB infection, an association study with 424 patients of fresh pulmonary tuberculosis (PTB) and 424 healthy controls was performed. Moreover, classification and regression tree and multifactor dimensionality reduction were employed to explore the effects of gene–gene interactions on cavitary PTB. The results indicated that both the heterozygous genotype GC and homozygous genotype CC in rs3809849 had significant effects on the risk of PTB (OR 1.42, 95 % CI 1.06–1.92, p 0.019; OR 1.55, 95 % CI 1.04–2.33, p = 0.033, respectively), and heterozygous genotype CT in rs9061 also had similar effects (OR 1.43, 95 % CI 1.07–1.90, p = 0.014). The rs3809849 and rs9905742 in MYBBP1A were also significantly associated with cavitary PTB (p = 0.00046 and 0.039, respectively), while rs9061 in SP110 had no such association (p = 0.06931) except its significant association with non-cavitary PTB (p = 0.0093). The interaction of MYBBP1A and RELA had significant effect on cavitary PTB (OR 4.24, 95 % CI 1.44–12.49, p = 0.005). These suggest that MYBBP1A instead of SP110 may be a genetic risk factor for cavitary PTB and play important effects on its whole progress.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With an estimated 8–9 million new cases occurring annually around the world, tuberculosis (TB) is still one of the major health issues worldwide [1]. Mycobacterium tuberculosis (MTB), responsible for developing clinical tuberculosis, can cause caseous necrosis in the host lung, which goes further into lung cavities through liquefaction. Pulmonary cavitation, the severe pathological consequence of pulmonary tuberculosis (PTB), is important clinical feature of high-level replicating or destructive MTB infection [2]. And lung cavities are known to serve as an excellent growth place for MTB. Therefore, the identification of host inner genetic factors for cavitary PTB is necessary for unveiling the progression and pathogenesis of tuberculosis in humans.
The SP110 gene has been shown in mice to have the ability to control MTB growth in macrophages and induce infected macrophage cells apoptosis [3]. The human SP110 gene is postulated to play roles in the development of clinical TB [4]. And current meta-analysis result supports that the SP110 rs9061 T allele may confer significant risk of PTB in East Asian populations [5]. However, SP110’s effects on progression to lung cavitation are unknown yet. Studying SP110’s effects on cavitary PTB is of help to know gene effects on the pathological progression of MTB infection.
In the present study, the effects of main single nucleotide polymorphisms (SNPs) in SP110 on cavitary PTB are examined. Furthermore, MYBBP1A and RELA, which are known to be SP110’s interacting proteins and be involved in SP110’s induction of macrophage apoptosis, are also studied [6, 7]. Since the sub-phenotype of complex disease may usually result from interactions of multiple genetic factors, multiple methods of classification and regression tree (CART) construction and multifactor dimensionality reduction (MDR) analysis are employed to detect the effects of gene–gene interactions on the cavitary PTB.
Materials and methods
Subjects
A total of 424 patients with pulmonary TB without previous episodes of TB were recruited from the Chongqing Daping Hospital and Shanghai Pulmonary Hospital in southern China from September 2010 to June 2013. All patients were HIV negative, and none presented with other infectious diseases or immunosuppressive conditions, and diagnosis of TB rested on chest radiographs and was confirmed by sputum smears and MTB culture. Severe form of TB was identified based on the appearance of cavities revealed by chest radiographs, denoting clinically destructive TB. Among the patients, 299 were men and 125 women; the mean age (±SD) was 40.08 ± 14.64 years. And among these patients, 159 had lung cavities and 265 had no lung cavities.
A total of 424 blood donors matched with cases were recruited as healthy controls described in the previous study [5], among whom, 284 were men and 140 women; the mean age (±SD) was 41.17 ± 17.68 years. There were no significant differences between PTB patients and healthy controls with regard to gender or age (p = 0.266 and 0.329, respectively). Healthy control subjects had negative tuberculin skin tests with indurations of <5 mm on the fourth day after inoculation, no history of TB and no evidence of prior TB noted in chest radiographies. This study was approved by the Ethics Committees of the Hospitals. And oral or written informed consent was provided by all subjects.
Genotyping analysis
Three tagging SNPs (rs3809849, rs10852863, rs9905742) in MYBBP1A, two tagging SNPs (rs9061, rs1135791) in SP110 and one tagging SNP (rs11820062) in RELA genes were selected as the first five SNPs and were found to be significantly associated with TB [5], and the last tagging SNP was located in the link disequilibrium region including whole RELA gene. All these SNPs were genotyped as previously described [5]. Briefly, all SNPs except rs9061 were genotyped using MASSarray platform (Sequenom; CA). The rs9061 and another SNP rs1135791 as an evaluator for the above genotyping method were tested using the SNaPshot Kit (Applied Biosystems; CA). For MASSarray, multiplex PCR was performed, and purified extension reaction products were spotted onto spectroCHIPs and determined by matrix-assisted laser desorption–ionization time-of-flight (MALDI-TOF) mass spectrometry. For SNaPshot method, PCR was firstly performed, and then, the products were then processed as templates for a mini-sequencing extension reaction using SNaPshot Kit. Finally, purified products were sequenced by capillary electrophoresis on an ABI3730xl DNA analyzer (Applied Biosystems; CA).
Statistical analysis
A Chi-squared test and t test were used to test the difference of gender or age between patients and controls. Fisher’s exact test was used to test Hardy–Weinberg equilibrium (HWE) for each SNP in control subjects. Odd ratios (ORs) and 95 % confidence intervals (CIs) for the heterozygous and homozygous carriers of the minor allele were calculated using the homozygous carrier of the major allele as the reference. The association between the genotypes of individual SNPs and PTB was determined using a Cochran–Armitage trend test. The data analysis was performed using the Stata 12.0 software (Stata Co., College Station, TX).
High-order gene–gene interactions were explored using CART analysis with CART 6.0 software (Salford Systems, USA). CART uses recursive partitioning to build a decision tree that enables the identification of subgroups of individuals at different levels of risks and with specific genotype interactions. Subgroups of individuals having different risk associations with TB were identified in the different terminal nodes of the tree, indicating the potential presence of interactions. Finally, the risk of these subgroups was evaluated by using logistic regression (LR) analysis. ORs and 95 % CIs were calculated with the least percentage of cases as the reference.
The free MDR software (http://www.multifactordimensionalityreduction.org/) was used to identify possible locus–locus and gene–gene interaction models, which were associated with cavitary PTB risk. The MDR is a nonparametric, model-free method for the detection and characterization of gene–gene interactions in small sample sizes [8]. Through dividing genotype factors into high and low risk level, MDR can effectively reduce the multifactor classification and prediction from high dimension to one dimension. In this study, Tuned ReliefF (TuRF) filter algorithm was applied to discard noisy SNPs and avoid overfitting. The best candidate interaction models for each locus were selected with the maximal testing balance accuracy (TBA) and the cross-validation consistency (CVC). The best interaction model was determined using 1,000-fold permutation testing, whose result was viewed statistically significant at the 0.05 level.
To visualize the MDR results, interaction entropy graphs containing nodes for corresponding SNPs and pairwise connections were constructed [9]. In the interaction entropy graphs based on information theory, synergy means positive interactions, while redundancy means negative interactions. Nodes for individual SNPs are connected in a pairwise way. The percentage of entropy removed by each SNP/pairwise connection is visualized for each node/line. And the red color represents a high degree of synergy with more entropy percentage, orange a lesser degree, and gold represents independence or a midway point between synergy and redundancy.
Results
Before testing the effects of each SNP on cavititary PTB, we tested the susceptibility to MTB infection of each SNP. Both the heterozygous genotype GC and homozygous genotype CC in rs3809849 were associated with increased risk of PTB (OR 1.42, 95 % CI 1.06–1.92, p = 0.019; OR 1.55, 95 % CI 1.04–2.33, p = 0.033, respectively), and heterozygous genotype CT in rs9061 (OR 1.43, 95 % CI 1.07–1.90, p = 0.014) was associated with increased risk of PTB (Supplementary Table 1).
Genotype distributions of rs3809849 and rs9905742 in the MYBBP1A gene were significantly associated with cavitary PTB (p = 0.00046 and 0.039, respectively), and SP110 rs9061 polymorphism was found to be associated with non-cavitary PTB (p = 0.0093; Supplementary Table 2). After the p value was adjusted using Bonferroni’s method, the heterozygous genotypes GC at rs3809849 and TA at rs9905742 were associated with increased risk of cavitary PTB (OR 1.9, 95 % CI 1.29–2.82, p = 0.006; OR 2.47, 95 % CI 1.37–4.46, p = 0.018, respectively (Table 1). Both the rs3809849 and rs9905742 polymorphisms were associated with increased risk to cavitary PTB in both the recessive and multiplicative genetic models (Supplementary Table 3).
Furthermore, with patients with cavitary PTB compared to the patients with non-cavitary PTB, the heterozygous genotypes GC at rs3809849 was associated with increased risk of lung cavitation during PTB (OR 1.69, 95 % CI 1.11–2.58, p = 0.015). However, after the p value was adjusted using Bonferroni’s method, no genotype of each SNP was found to be significantly associated with lung cavitation formation (Table 2).
The up to four orders of best interaction models from MDR analysis were shown in Table 3, along with their CVCs and TBAs for data sets of cavitary TB. The best one-factor model for predicting cavitary PTB risk was rs3809849 (CVC = 10/10, TBA = 0.5782, p = 0.027). The best model turned out to be the three-locus model comprising of rs3809849, rs1135791 and rs11820062 with a CVC of 10/10 and TBA equals to 0.6344 (p < 0.001). While the two-locus model had lower TBA (56.76 %), and four-locus models had lower CVC (9/10) than the three-locus model.
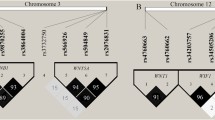
Consistent with the MDR best one-factor model, the initial split of the root node was rs3809849 (Fig. 1 ), suggesting that this SNP was the strongest risk factor for cavitary PTB among the examined SNPs. Using terminal node two with the least percentage of cases as the reference, among individuals carrying the rs3809849 C allele, the maximum risk was observed for terminal node 4 with the rs11820062 CT or TT genotypes (OR 4.24, 95 % CI 1.44–12.49, p = 0.0050).

Classification and regression tree (CART) analysis of MYBBP1A, SP110 and RELA gene polymorphism in case of cavitary PTB. Class 0 refers to the healthy controls corresponding to the red bar, and class 1 refers to the TB patients corresponding to the blue bar (color figure online)
Interaction entropy graphs were constructed using the MDR results obtained from the analysis of cavitary PTB data set. In cavitary PTB, rs3809849 tended to have a strong independent effect because the entropy removing this individual SNP was 1.55 % in a high degree, and a strong synergistic interaction was found between rs1135791 and rs11820062 as the combination had the highest entropy of 2.72 % among the entropy for SNPs or connections (Supplementary Fig. 1).
Discussion
TB, usually caused by MTB, is a re-emerging disease epidemic in many parts of the world. Pulmonary cavitation, which is an ideal place for high-level replicating or destructive MTB infection, is the severe pathological phenotype of PTB [2]. The SP110 gene is emerging as a promising candidate for controlling MTB infections in mice model [3]. The meta-analysis result suggests that the SP110 rs9061 T allele may confer risk of PTB in East Asian populations [5]. The previous study results also demonstrate that both Mybbp1A and sp110 may be a susceptible gene for PTB [5]. However, in that study, the patients with previous episodes of TB might be included, which could not sufficiently explain the genetic factors determining the susceptibility of fresh MTB infection. Moreover, SP110 and its associated genes’ effects on progression to lung cavitation are unknown yet. Thus, we examined the effect of each SNP and the interaction of gene–gene on cavitary PTB.
In the present study, it was found that the presence of the genotypes CT at rs9061 in SP110, and GC and CC at rs3809848 in MYBBP1A independently conferred susceptibility to PTB. It suggested that MYBBP1A not SP110 tended to play an important role in elevating the risk of developing cavitary PTB. MTB, an ancient microbiology, has sophisticated strategies to cope with host monocytic cells. After entering a host organism, MTB can actively induce lung epithelioid cell granulomas and subsequently necrosis or stay in macrophages as dormant bacteria. The morphological hallmark of necrosis is the appearance of a cheese-like material termed caseous necrosis. Caseation may lead to extensive tissue damage, such as liquefaction and formation of cavities. Within the cavities, MTB can proliferate extracellularly in a larger scale and prepare to spread throughout the body or release in aerosols [10]. In human, following induction by interferon-α or interferon-γ, SP110 becomes manifest as a leukocyte-specific component of the nuclear body, which has been reported to inhibit virus replication [11, 12]. The SP110’s homologue in mice has been proved to have the ability to control MTB growth in macrophages and induce infected macrophage cells apoptosis [3]. In current study, SP110 gene has been found to be associated with the PTB, while not to be associated with the presence of pulmonary cavities. It is reasonable to speculate that during TB active infection, SP110 is induced and plays important roles to intracellularly resist TB through inducing apoptosis of infected macrophages. Usually, the apoptosis of macrophages means fewer cells acting as protected MTB sanctuaries. However, sometimes, the apoptosis of macrophages is of help for MTB to elude immune system surveillance and spread surviving bacilli [13]. Thus, the association between the SP110 and PTB might be due to decreased replication of the bacilli in the macrophage rather than to extracellular spread of surviving bacilli and lung cavitation.
It should be noted that genotype distribution of each SNP is not found to be significantly different between cavitary PTB patients and non-cavitary PTB ones. Although the heterozygous genotype GC in rs3809849 is associated with lung cavity formation (p = 0.015), after adjustment using Bonferroni’s method, no genotype of each SNP was found to be significantly associated with lung cavitation formation. Thus, the MYBBP1A interpreted as a predisposed risk factor to cavitary PTB might be not via the effects on the lung cavitation formation. However, given the possibility of most non-cavitary PTB patients with other severe features, genetic analyses between patients with and without cavitary PTB would not obtain real results. And thus, the comparison between patients with cavitary PTB and healthy controls is proper.
Furthermore, we performed multiple methods including MDR and CART, which could identify many genes interactions based on genetic data. Compared with traditional parametric statistical methods, such as logistic regression analysis, MDR analysis, which uses a nonparametric/model-free approach to detect nonlinear interactions among discrete genetic factors, can more easily characterize the complex multifactor interactions with the sparseness of the data in certain dimensions and of the insufficient power. The results reflect that rs3809849 in MYBBP1A has strong association with cavitary PTB as being the only one factor in the best MDR model and the first split in CART. Both CART and MDR results suggest there is the interaction between the rs3809849 polymorphism in MYBBP1A with rs11820062 polymorphism in RELA. MYBBP1A is a transcription factor critical for hematopoietic cell proliferation and differentiation. This finding is consistent with that MYBBP1A can bind to RELA in Jurkat T cells [7]. RELA is a member of the NF-κB family of structurally related inducible transcription factors [14]. NF-κB, which is known to interact with IFN-γ during TB development, regulates the expression of a wide variety of human genes, including factors regulating apoptosis [15, 16]. This interaction might be a potential risk factor for cavitary PTB.
There may be some limits to the current study. The sample size of current study is not large. However, based on the effect size of 0.15, the minimum total sample size needed is 732 at 5 % level of significance with a power value of 90 %. A total of 848 samples in the present study can meet this criterion. And the environment risk factors, such as smoking, are not studied. Smoking is well known to exacerbate most lung diseases, and the present study provides only genetic information about the genes involved in cavitary PTB.
The percentage of pulmonary TB cases suffer from cavitary disease varies significantly by age and other risk factors from 50 to 80 % [1]. However, PTB patients with cavitary disease are more contagious than those without and have increased risk of relapse. The current study suggests that MYBBP1A may be a genetic risk factor for cavitary PTB, and hints that when treating patients with cavitary PTB, physicians may need pay extra attention on the genetic factors and provide individual therapy.
Conclusion
In conclusion, MYBBP1A and SP110 may be genetic risk factors for TB in the Han Chinese population. MYBBP1A rather than SP110 may play important roles in predisposing risk to cavitary PTB.
Abbreviations
- MTB:
-
Mycobacterium tuberculosis
- MYBBP1A:
-
MYB-binding protein
- MDR:
-
Multifactor dimensionality reduction
- CART:
-
Classification and regression tree
- LR:
-
Logistic regression
References
WHO. Global tuberculosis control: WHO report 2011. Avenue: World Health Organization. 2011.
Sakamoto K. The pathology of Mycobacterium tuberculosis infection. Vet Pathol. 2012;49:423–39.
Pan H, Yan BS, Rojas M, et al. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 2005;434:767–72.
Tosh K, Campbell SJ, Fielding K, et al. Variants in the SP110 gene are associated with genetic susceptibility to tuberculosis in West Africa. Proc Natl Acad Sci USA. 2006;103:10364–8.
Cai L, Deng SL, Liang L, et al. Identification of genetic associations of SP110/MYBBP1A/RELA with pulmonary tuberculosis in the Chinese Han population. Hum Genet. 2013;132:265–73.
Cai L, Pan H, Trzcinski K, Thompson CM, Wu Q, Kramnik I. MYBBP1A: a new Ipr1’s binding protein in mice. Mol Biol Rep. 2010;37:3863–8.
Owen HR, Elser M, Cheung E, Gersbach M, Kraus WL, Hottiger MO. MYBBP1a is a novel repressor of NF-kappa B. J Mol Biol. 2007;366:725–36.
Hahn LW, Ritchie MD, Moore JH. Multifactor dimensionality reduction software for detecting gene–gene and gene–environment interactions. Bioinformatics. 2003;19:376–82 (Oxford, England).
Jakulin ABI. Analyzing attribute dependencies. In: Lecture Notes in Computer Science, vol 2838; 2003. p. 229–40.
Dannenberg AM Jr. Delayed-type hypersensitivity and cell-mediated immunity in the pathogenesis of tuberculosis. Immunol Today. 1991;12:228–33.
Bloch DB, Nakajima A, Gulick T, et al. Sp110 localizes to the PML-Sp100 nuclear body and may function as a nuclear hormone receptor transcriptional coactivator. Mol Cell Biol. 2000;20:6138–46.
Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. BioEssays. 2004;26:963–77.
Bocchino M, Galati D, Sanduzzi A, Colizzi V, Brunetti E, Mancino G. Role of mycobacteria-induced monocyte/macrophage apoptosis in the pathogenesis of human tuberculosis. Int J Tuberc Lung Dis. 2005;9:375–83.
Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8.
Chan ED, Morris KR, Belisle JT, et al. Induction of inducible nitric oxide synthase-NO* by lipoarabinomannan of Mycobacterium tuberculosis is mediated by MEK1-ERK, MKK7-JNK, and NF-kappaB signaling pathways. Infect Immun. 2001;69:2001–10.
Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66.
Acknowledgments
The authors thank all study participants for their donation of blood samples. This work was supported by the National Infectious Disease Prevention and Cure Special Project (No. 2013ZX10003006-003-002), the National Nature Science Foundation of China (No. 31101015), the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (No. 81421061) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (no.12Z102050009).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Shi-Yao Jiang and Lin-Lin Li have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jiang, SY., Li, LL., Yue, J. et al. The effects of SP110’s associated genes on fresh cavitary pulmonary tuberculosis in Han Chinese population. Clin Exp Med 16, 219–225 (2016). https://doi.org/10.1007/s10238-015-0339-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-015-0339-4