
Abstract
PCDH10 is a key tumor suppressive gene for nasopharyngeal, esophageal, and other carcinomas with frequent methylation. In this study, we investigated the potential epigenetic modification of the PCDH10 gene by hepatitis B virus × protein (HBx), a pivotal factor in the progression of HBV replication and potential carcinogenesis. PCDH10 expression was found to be down-regulated in 9/13 (69.2 %) of hepatocellular carcinoma (HCC) cell lines. Decreased PCDH10 expression was correlated with the methylation status of the PCDH10 promoter. Treatment with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (Aza) was sufficient to restore PCDH10 mRNA expression by suppressing PCDH10 promoter methylation in HepG2 cells. Treatment with Trichostatin A alone had no significant effect on PCDH10 expression but enhanced the effect of Aza. PCDH10 methylation was further detected in 76 % (38 of 50) of HCC tissues compared with 40 % (20 of 50) of paired adjacent tissues, with no methylation detected in normal human liver tissues. There were significant correlations between methylation status of PCDH10 and tumor size, serum AFP levels, metastasis or TNM staging (P < 0.05). Moreover, PCDH10 promoter methylation status was not associated with HBV infection in our panel of 50 primary HCC tumors, and transfection with HBX could not alter the status of PCDH10 promoter methylation. Collectively, these observations suggested that the expression of PCDH10 was silenced in HCC via de novo DNA methylation independent of HBV infection or HBX expression, and PCDH10 might form a potentially useful therapeutic target for HCC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is characterized by late detection and fast progression, and it is believed that epigenetic disruption may be the cause of its molecular and clinicopathological heterogeneity. A better understanding of the global deregulation of methylation states and how they correlate with disease progression will aid in the design of strategies for earlier detection and better therapeutic decisions.
Human hepatitis B virus (HBV) infection is a leading cause of HCC in China [1]. HCC arises as a result of genetic and epigenetic abnormalities. Interestingly, several studies have reported a strong correlation between HBV infection and epigenetic alteration of many tumor-suppressor genes, such as p16 [2] and GSPT1 [3], whose expressions were frequently down-regulated in HCC by promoter hypermethylation.
The HBV × protein (HBx), a pivotal factor in the progression of HBV infection [4], replication [5], pathogenesis, and potential carcinogenesis [6], has been implicated as a potential trigger of the epigenetic deregulation of some key TSG genes [2, 3]. Recent study by Park et al. [7] has further demonstrated that HBx can interact directly with and recruit the de novo DNA methyltransferase DNMT3a to the regulatory promoters of interleukin-4 receptor and metallothionein-1F and subsequently silenced their transcription via de novo DNA methylation. Moreover, the transcriptional levels of the target genes in HCC specimens were strongly correlated with the occurrence of HBx, providing an alternative mechanism within HBx-mediated transcriptional repression.
PCDH10 is recently demonstrated as a key tumor suppressive gene (TSG) for colorectal, nasopharyngeal, esophageal, hepatocellular, breast, cervical, gastric, lung, hematologic malignancies [8], and prostate cancers with frequent methylation [9], and it has reported to be involved in proliferation inhibition, apoptosis induction, and invasion repression [10], however, its role in hepatocellular carcinogenesis is still largely unknown.
In this study, we used methylation-specific PCR (MSP) and bisulfite genomic sequencing (BGS) to explore the methylation status of the upstream PCDH10 promoter from HepG2 cells, its HBX transfectants, HCC patients and adjacent non-cancerous tissues in comparison with normal liver tissues where neither viral infection nor hepatitis existed. We demonstrated that PCDH10 expression was significantly down-regulated in both HCC cell lines and HCC patient samples through de novo DNA methylation, which could partially reversed upon inhibition of DNMTs by Aza in HepG2 cells and its HBX transfectants. However, the methylation status of the PCDH10 gene in HCC was not affected by HBV infection or HBX expression.
Materials and methods
HCC cell lines
Thirteen HCC cell lines (SNU398, HBV+; SNU449, HBV+; SNU475, HBV+; SNU387, HBV+; SNU423, HBV+; Hep3B, HBV+; HepG2, HBV−; HUH1, HBV+; HUH4, HBV+; HUH6, HBV−; HUH7, HBV−; Mahlavu, HBV−; PLC/PRF/5, HBV+) were obtained from Dr. Qian Tao (Cancer Epigenetics Laboratory, Department of Clinical Oncology, the Chinese University of Hong Kong, Shatin, NT, Hong Kong). Cell lines were maintained in RPMI1640 medium (Gibco BRL, Rockville, MD, USA) with 10 % fetal bovine serum (Gibco BRL).
Patients and the collection of the liver tissue specimens
Diagnostic criteria for HCC included liver biopsy or radiographic evidence consistent of HCC as defined by accepted guidelines [11]. A total of 50 paired primary HCC tissues and adjacent non-cancerous tissues were obtained during surgical resection of HCC patients with no preoperative chemotherapy or radiotherapy according to a standard protocol. The adjacent non-cancerous tissues were subsequently verified by histology to be free of tumor infiltration and the adjacent non-cancerous specimens with tumor cell infiltration were excluded. All the specimens were snap-frozen in liquid nitrogen and stored at −80 °C for molecular analyses. The remaining tissue specimens were fixed in 10 % formalin and embedded in paraffin for routine histological examination. All tissue specimens for this study were obtained according to protocols approved by the Medical Ethics Committee on human research in the First Affiliated Hospital of Chongqing Medical University, and written informed consent was obtained from all the participants before enrollment. The clinicopathological features of the patients were shown as in Table 1. Tumor was staged according to the TNM staging system. All histological assessments were made by an experienced pathologist.
Bisulfite modification of DNA
Bisulfite induces deamination of unmethylated cytosines, converting unmethylated CpG sites to UpG without modifying methylated sites, allowing their differentiation by methylation-specific polymerase chain reaction (MSP) or BGS. Genomic DNA was extracted from HCC liver tissues or HCC cell lines using DNA Mini Kit (Qiagen, Valencia, CA, USA) and treated with sodium bisulfite using a Zymo DNA modification kit (Zymo Research, Orange, CA, USA).
Reverse-transcription PCR (RT-PCR)
Total RNA was extracted from HCC specimens or HCC cell lines by SV Total RNA Kit (Promega, CA, USA). The messenger RNA (mRNA) expression level of PCDH1O was determined by RT-PCR using primers 5′-GGTGCTGTACGTGAACGAGA-3′ and 5′-CACCTTTAGAGACTCGCG-3′. Primers for GAPDH (5′-GAGTCAACGGATTTGGTCGT-3′ and 5′-TTAGGGTAGTGGTAGAAGGT-3′) were included as loading control.
Demethylation by DNA demethylating agent 5-aza-2-deoxycytidine (Aza)
HepG2 cells were seeded at a density of 3.0 × 104 cells/well, and after 48 h, cells were treated with 5 µM of the DNA demethylating agent 5-Aza (Sigma–Aldrich, St. Louis, MO, USA) for 48 h with or without 500 nM Trichostatin A (TSA) for 24 h. Cells were then harvested for DNA and RNA extractions.
Methylation-specific PCR (MSP)
Methylation-specific PCR (MSP) was performed as described previously [10, 12]. Briefly, MSP was performed for 40 cycles using AmpliTaq Gold and hot-start with the following primers: methylation-specific, PCDH10m1: 5′-TCGTTAAATAGATACGTTACGC, PCDH10m2: 5′-TAAAAACTAAAAACTTTCCGCG, or unmethylation-specific, PCDH10u1:5′-GTTGTTAAATAGATATGTTATGT, PCDH10u2: 5′-CTAAAAACTAAAAACTTTCCACA.
Bisulfite genomic sequencing (BGS)
For BGS, 1 µl of bisulfite-treated DNA was amplified using primers BGS1: 5′-GTTGATGTAAATAGGGGAATT and BGS2: 5′-CTTCAACCTCTAAACCTATAA. The PCR products were cloned into PMD18-T Vector (Takara). Ten colonies were chosen randomly for plasmid DNA extraction with Qiaprep Spin Mini kit (Qiagen, Valencia, CA, USA) and were sequenced. Sequencing analysis was performed by SeqScape software (Applied Biosystems, Foster City, CA, USA).
Statistical analysis
The difference of PCDH10 mRNA expression between tumor and adjacent non-cancerous tissues was analyzed by the Mann–Whitney U test. The χ2 test was employed for the comparison of patient characteristics and distributions of PCDH10 methylation. All analyses were performed using SAS for Windows software, version 9 (SAS Institute, Inc., Cary, NC, USA). Two-tailed P values <0.05 were considered statistically significant.
Results
Methylation of PCDH10 CpG islands (CGI) contributes to the down-regulation of PCDH10 mRNA expression in HCC cell lines
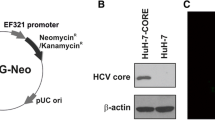
PCDH10 mRNA expression was found to be down-regulated in 9/13 (69.2 %) of the HCC cell lines (Fig. 1a). We hypothesized that the low level of PCDH10 mRNA expression in HCC cells could reflect epigenetic silencing through hypermethylation of its promoter, a common mechanism of inactivating tumor-suppressor genes in cancer. We thus further analyzed the methylation status of the PCDH10 CGI with MSP in HepG2 cells in which PCDH10 expression was found to be totally silenced. As expected, the PCDH10 CGI was methylated in HepG2 cells (Fig. 1b).

Silencing of PCDH10 by promoter methylation in HCC cell lines. a mRNA expression of PCDH10 in different cell lines and normal liver cells, GAPDH was used as a control; b methylation status and expression of PCDH10 in HepG2 cells; c methylation status and expression of PCDH10 in HepG2 cells transfected with HBx. Lane 1 HepG2; lane 2 HepG2 transfected with HBx (HepG2–HBx); lane 3 HepG2 vector control
We further examined the methylation status of the PCDH10 CGI by high-resolution bisulfite genome sequencing (BGS) analysis of 36 CpG sites within the CGI, including those CpG sites analyzed by MSP. Nearly, all CpG sites examined were methylated in HepG2 cells (Fig. 2c).

Pharmacologic demethylation activated PCDH10 expression in silenced HepG2 cells. a mRNA expression of PCDH10 in HepG2 cells treated with AZA/TSA; b methylation status of PCDH10 in HepG2 cells treated with AZA/TSA; c High-resolution methylation mapping of CpG sites by BGS confirmed the pharmacologic demethylation of PCDH10 in HepG2 cells
Activation of PCDH10 expression by pharmacological demethylation
To further determine whether CGI methylation directly mediates PCDH10 silencing, we compared the PCDH10 expression levels in HepG2 cells before and after treatment with the DNA methyltransferase inhibitor 5-Aza), together with or without histone deacetylase inhibitor TSA. Aza alone was sufficient to restore PCDH10 mRNA expression in HepG2 cells. Treatment with TSA alone had no significant effect on the expression of PCDH10 but enhanced the effect of Aza (Fig. 2a). Meanwhile, both MSP and BGS showed that the CGI was dramatically demethylated in the presence of the drug (Fig. 2b, c), revealing a direct link between PCDH10 silencing and CpG methylation. These results confirmed that PCDH10 down-regulation was directly mediated by CGI methylation, and Aza alone was sufficient to restore PCDH10 mRNA expression by suppressing PCDH10 promoter methylation in HepG2 cells.
Methylation of the PCDH10 CGI in primary carcinomas
We further analyzed the expression level of PCDH10 mRNA and the methylation status of PCDH10 in a large collection of primary HCC samples. PCDH10 mRNA expression was found to be repressed or down-regulated in 32/50 (64 %) of the HCC tissues compared with 30 % (15 of 50) of paired adjacent tissues (P < 0.01) and none in normal liver tissues (Fig. 3a). Concordantly, PCDH10 methylation was detected in 76 % (38 of 50) of HCC tissues compared with 40 % (20 of 50) of paired adjacent tissues (P < 0.0001) and none in normal liver tissues (Fig. 3b, c), highlighting the importance of tumor-specific PCDH10 methylation in hepatocellular carcinogenesis.

Methylation of the PCDH10 CGI in primary carcinomas. a mRNA expression of PCDH10 in HepG2 cells in tumor tissues, adjacent tissues and normal tissues; b methylation status of PCDH10 in tumor tissues, adjacent tissues and normal tissues; c high-resolution methylation mapping of CpG sites by BGS in tumor tissues and normal tissues
Methylation of the PCDH10 CGI in HCC cell lines and primary carcinomas was independent of HBV infection or HBX expression status
Among all the HCC cell lines detected, 9 were reported to be positive for HBV infection; however, only 5 were found to express low levels of PCDH10 mRNA (Fig. 1a). Transfection of HepG2 cells with HBX protein could not alter PCDH10 promoter methylation and PCDH10 expression (Fig. 1c). In addition, chronic HBV infection was detected in 23 of 50 (46 %) HCC tissues, and only 17 was detected to have PCDH10 methylation (Table 1). PCDH10 methylation was not associated with HBV infection in HCC tissues or HBX expression status in HCC cell lines (Fig. 4).

Silencing of PCDH10 by promoter methylation in HBV negative/positive HCC tissues. a mRNA expression of PCDH10 in HBV negative/positive HCC tissues; b methylation status of PCDH10 in HBV negative/positive HCC tissues
Association between the PCDH10 methylation status and the clinicopathological characteristics of HCC patients
The association between clinicopathological features and the PCDH10 methylation status in human HCC was listed as in Table 1. Though there was no correlation between the methylation status of PCDH10 and clinicopathological features such as age, sex, chronic HBV infection, ethanol abuse, and cirrhosis, significant correlations were indeed found between the methylation status of PCDH10 and tumor size, serum AFP levels, metastasis, and TNM staging (P < 0.05).
Discussion
In the present study, loss of the PCDH10 mRNA expression in HepG2 cells was found to be correlated well with the methylation of the upstream PCDH10 promoter. 5′-Aza treatment re-induced the PCDH10 mRNA expression in both HepG2 and HepG2–HBX transfectants with concomitant demethylation of the upstream PCDH10 promoter. Upstream PCDH10 promoter was further found to be frequently hypermethylated in primary HCC liver samples with concomitant decreased PCDH10 mRNA expression. Though there was no correlation between the methylation status of PCDH10 and clinicopathological features such as age, sex, chronic HBV infection, ethanol abuse and cirrhosis, well correlations between the methylation status of PCDH10 and tumor size, serum AFP levels, metastasis, and TNM staging were observed. Taken together, these results suggest that epigenetic inactivation of PCDH10 might be an important step in hepatocellular carcinogenesis.
PCDH10 has been reported to be a functional TSG, and its transcriptional silencing and promoter methylation were frequently detected in a variety of carcinomas [8, 10, 12]. Here, we further detected its frequent tumor-specific methylation in HCC cell lines and primary tumor tissues, which extended previous work on CDH10 to gastric cancer [10]. Methylation treatment and methylation analyses indicate that promoter methylation is the principal regulatory mechanism of reduced PCDH10 expression in HCCs. However, there are unmethylated alleles in some HCC tissues with no PCDH10 mRNA expression detected or methylated alleles in some HCC tissues with normal PCDH10 mRNA expression, suggesting other regulating mechanisms including histone modification, microRNA or transcriptional repressors and other TSG rather than PCDH10 also contribute to the pathogenesis of HCC.
PCDH10 belongs to the protocadherin superfamily and it is widely expressed in normal tissues [12–14]. It has been reported that PCDH10 mediated tumor-suppressor effect in cancer cells via induction of apoptosis, control of cell growth, and inhibition of cell invasion and metastasis [12, 15, 16]. While PCDH10 is frequently silenced by methylation in HCC, ectopic expression of PCDH10 may suppress tumor cell growth, migration, invasion and colony formation. In this study, we also found that pharmacologic demethylation of PCDH10 by Aza treatment could restore the expression of methylation-silenced gene expression, and this strategy may be potentially explored as a HCC therapeutic strategy through reactivating the biological functions of PCDH10.
Hepatocellular carcinoma (HCC) varies greatly in clinical outcomes depending on the progress of individual tumors [17, 18]. We further analyzed the clinical significance of PCDH10 promoter methylation and its associations with clinical parameters in HCC patients. There were significant correlations between the methylation status of PCDH10 and tumor size, serum AFP levels, metastasis or TNM staging in patients, suggesting that PCDH10 methylation in HCC patients may be further explored as a new biomarker for monitoring disease progression or as a prognostic factor for relapse in HCC patients with PCDH10 methylation. However, further studies are required to clarify the significant association between the PCDH10 methylation and the progression of tumoral process.
Chronic infection with HBV is a major risk of HCC, it has been shown that most HBV-related HCC is integrated with viral DNA [19]. Several previous studies have demonstrated that HBV infection was associated with epigenetic alteration of some tumor-suppressor genes, including p16 and GSPT1 [2, 3]. However, here, we found that HBV infection was not related to PCDH10 methylation in HCC tissues, and there was no significant correlation between serum HBsAg concentrations or HBV–DNA copies and PCDH10 methylation status (data not shown). HBx is a small 17-kDa soluble protein in the nucleus and cytoplasm of host cells, which is known to play an essential role in HBV-induced live carcinogenesis [20]. Nuclear HBx could affect gene transcription by interacting with a specific transcriptional machinery at the promoter level [21]. However, transfection of HepG2 cells with HBx could not alter PCDH10 methylation status and its mRNA expression. Our results therefore suggest that HBV infection or HBX protein was not involved in the methylation of the PCDH10 CGI in HCC cell lines and primary HCC tissues.
In summary, silencing of PCDH10 in HCC via de novo DNA methylation independent of HBV infection or HBX protein expression was investigated in this study. Because epigenetic inactivation of PCDH10 might be an important step in hepatocellular carcinogenesis, targeting PCDH10 by pharmacologic demethylation may have important therapeutic implications for HCC treatment, and its clinical exploration could be further investigated in animal model of HCC.
References
Fung J, Lai CL, Yuen MF (2009) Hepatitis B and C virus-related carcinogenesis. Clin Microbiol Infect 15:964–970
Jicai Z, Zongtao Y, Jun L et al (2006) Persistent infection of hepatitis B virus is involved in high rate of p16 methylation in hepatocellular carcinoma. Mol Carcinog 45:530–536
Zondervan PE, Wink J, Alers JC (2000) Molecular cytogenetic evaluation of virus-associated and non-viral hepatocellular carcinoma: analysis of 26 carcinomas and 12 concurrent dysplasias. J Pathol 192:207–215
Neuveut C, Wei Y, Buendia MA (2010) Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol 52:594–604
Gearhart TL, Bouchard MJ (2010) Replication of the hepatitis B virus requires a calcium-dependent HBx-induced G1 phase arrest of hepatocytes. Virology 407:14–25
De Mitri MS, Cassini R, Bernardi M (2010) Hepatitis B virus-related hepatocarcinogenesis: molecular oncogenic potential of clear or occult infections. Eur J Cancer 46:2178–2186
Park IY, Sohn BH, Yu E (2007) Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus × protein. Gastroenterology 132:1476–1494
Ying J, Gao Z, Li H et al (2007) Frequent epigenetic silencing of protocadherin 10 by methylation in multiple haematologic malignancies. Br J Haematol 136:829–832
Li Z, Li W, Xie J et al (2011) Epigenetic inactivation of PCDH10 in human prostate cancer cell lines. Cell Biol Int 35:671–676
Yu J, Cheng YY, Tao Q et al (2009) Methylation of protocadherin 10, a novel tumor suppressor, is associated with poor prognosis in patients with gastric cancer. Gastroenterology 136:640–651
Davila JA, Henderson L, Kramer JR et al (2011) Utilization of surveillance for hepatocellular carcinoma among hepatitis C virus-infected veterans in the United States. Ann Intern Med 154:85–93
Ying J, Li H, Seng TJ et al (2006) Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene 25:1070–1080
Yu B, Yang H, Zhang C et al (2010) High-resolution melting analysis of PCDH10 methylation levels in gastric, colorectal and pancreatic cancers. Neoplasma 57:247–252
Rosenbauer F, Owens BM, Yu L et al (2006) Lymphoid cell growth and transformation are suppressed by a key regulatory element of the gene encoding PU.1. Nat Genet 38:27–37
Li Z, Xie J, Li W et al (2011) Identification and characterization of human PCDH10 gene promoter. Gene 475:49–56
Cheung HH, Lee TL, Davis AJ et al (2010) Genome-wide DNA methylation profiling reveals novel epigenetically regulated genes and non-coding RNAs in human testicular cancer. Br J Cancer 102:419–427
Park JW, Finn RS, Kim JS et al (2011) Phase II, open-label study of brivanib as first-line therapy in patients with advanced hepatocellular carcinoma. Clin Cancer Res 17:1973–1983
Tanaka S, Arii S (2011) Molecular targeted therapy for hepatocellular carcinoma in the current and potential next strategies. J Gastroenterol 46:289–296
Yang JD, Roberts LR (2010) Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol 7:448–458
Keng VW, Tschida BR, Bell JB et al (2011) Modeling hepatitis B virus X-induced hepatocellular carcinoma in mice with the Sleeping Beauty transposon system. Hepatology 53:781–790
Xiang WQ, Feng WF, Ke W et al (2011) Hepatitis B virus × protein stimulates IL-6 expression in hepatocytes via a MyD88-dependent pathway. J Hepatol 54:26–33
Acknowledgments
This work was supported in part by the National Science Foundation of China [Grant Nos. 81071621 and 30973378], the Natural Science Foundation of Chongqing, China [Grant No. CSTC, 2010BB5390], the Science Foundation of Chongqing Municipal Bureau of Health [Grant No. 2010-2-090] and the Medical Science Foundation of the First Affiliated Hospital of Chongqing Medical University [Grant No. YXJJ 2009-12].
Conflict of interest
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Song Fang and Shi-feng Huang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Fang, S., Huang, Sf., Cao, J. et al. Silencing of PCDH10 in hepatocellular carcinoma via de novo DNA methylation independent of HBV infection or HBX expression. Clin Exp Med 13, 127–134 (2013). https://doi.org/10.1007/s10238-012-0182-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-012-0182-9